
Abstract
Objectives
Primary tumor tissue is often analyzed to search for predictive biomarkers and DNA-guided personalized therapies, but there is an incomplete understanding of the discrepancies in the genomic profiles between primary tumors and metastases, such as liver and lung metastases.
Methods
We performed in-depth targeted next-generation sequencing of 520 key cancer-associated genes for 47 matched primary and metastatic tumor samples which were retrospectively collected.
Results
A total of 699 mutations were detected in the 47 samples. The coincidence rate of primary tumors and metastases was 51.8% (n = 362), and compared to patients with liver metastases, patients with lung metastases had a significantly greater coincidence rate (P = .021). The number of specific mutations for the primary tumors and liver and lung metastases was 186 (26.6%), 122 (17.5%), and 29 (4.1%), respectively. Analysis of a patient with all three occurrences, including a primary tumor, liver metastasis, and lung metastasis, indicated a possible polyclonal seeding mechanism for liver metastases. Remarkably, multiple samples from patients with primary and metastatic tumors supported a mechanism of synchronous parallel dissemination from primary tumors to metastatic tumors that were not mediated through pre-metastatic tumors. We also found that the PI3K-Akt signaling pathway significantly altered lung metastases compared to matched primary tumors (P = .001). In addition, patients with mutations in CTCF, PIK3CA, or TP53 and LRP1B, AURKA, FGFR1, ATRX, DNMT3B, or GNAS had larger primary tumor sizes and metastases, especially patients with both LRP1B and AURKA mutations. Interestingly, CRC patients with TP53-disruptive mutations were more likely to have liver metastases (P = .016).
Conclusion
In this study, we demonstrate significant differences in the genomic landscapes of colorectal cancer patients based on the site of metastasis. Notably, we observe a larger genomic variation between primary tumors and liver metastasis compared to primary tumors and lung metastasis. These findings can be used for tailoring treatments based on the specific metastatic site.
Keywords
Introduction
Colorectal cancer (CRC) is the third most commonly diagnosed cancer, and the second most deadly cancer with 9.4% of cancer-related deaths, accounting for 935 000 cancer-related deaths worldwide in 2020. 1 Based on the American Cancer Society statistics, the 5-year relative survival ranging from 91% for patients diagnosed with localized CRC to 14% for patients diagnosed with distant CRC in the five major racial and ethnic group by all ages during 2012-2018. 2 A large-scale comprehensive integrative analysis has characterized somatic alterations in CRC3,4 without examining the different metastases. Most molecular comparisons have been made between primary tumors and liver metastases without examining lung metastases.5–7 The studies have revealed extensive intra-tumor heterogeneity (ITH), and therapeutic success depends on identifying major differences between the genomic profiles of metastases and primary tumors.8,9 Multi-region whole-exome sequencing has demonstrated ITH in primary tumors 10 and in matched primary and metastatic tumors in CRC. 11 To date, in-depth analyses of large series of colorectal cancer metastases are studied using either Whole-exome sequencing (WES) or Whole genome sequencing(WGS).12–14 Although these studies yielded extensive knowledge molecular landscape on the primary or metastatic colorectal cancer tumors, they do not specially reflect its molecular landscape discrepancies between liver and lung metastases. In addition, the study which was reported in detail on WES and WGS data of colorectal metastases included only 12 metastatic colorectal cancer patients (including lymph node metastases, liver metastases and omentum metastases), 15 the other studies which have characterized the mutational landscape of primary tumors and matched metastases in colorectal cancer patients using targeted next-generation sequencing (NGS) also used a limited panel of up to 182 genes in 13 patients(12 liver metastases and 1 peritoneal metastasis) 16 or a 48 gene TruSeq Amplicon Cancer PanelTM in 14 patients. 17 Therefore, genomic landscapes of primary and metastatic tumors and different genomic mutation profiles between liver and lung metastatic tumors remain insufficiently explored.
In this study, we comprehensively compared the genetic profiles of 520 genes in 47 samples from 18 CRC patients using NGS. Analysis of significant effects on signaling pathways, tumor mutational burden (TMB) and meaningful specific mutated genes in primary and metastatic tumor samples emphasized the clonal heterogeneity of CRC. We aimed to identify genetic differences between primary CRC tumors and associated metastases to the liver or lung, which are crucial to identify metastasis-specific molecular biomarkers and improve therapeutic approaches for CRC metastasis.
Materials and Methods
Patient Selection and Criteria
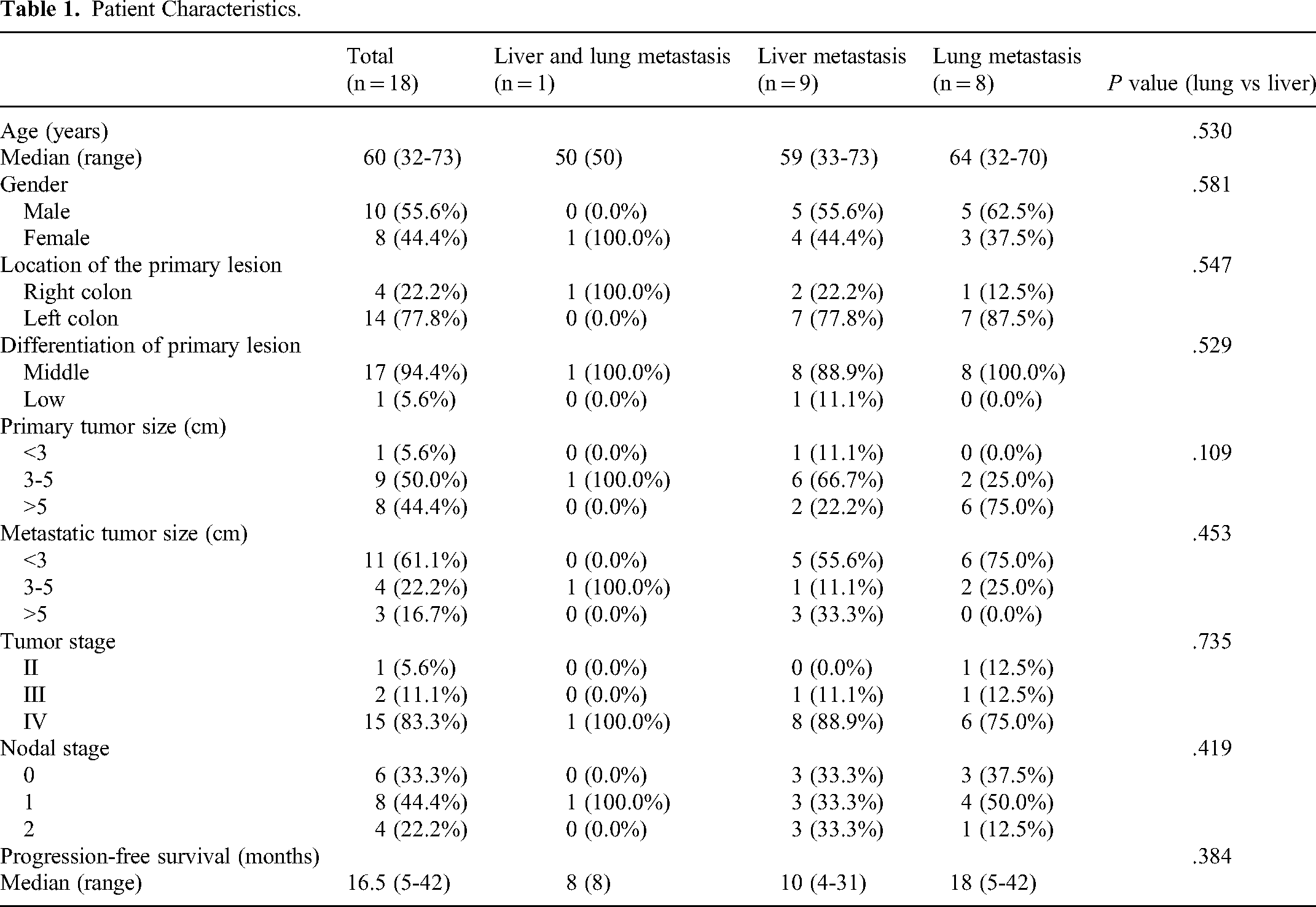
The study was a single center retrospective study, included 47 samples from 18 CRC patients. Nine patients had liver metastases, eight patients had lung metastases and one patient had synchronous liver and lung metastases. Clinico-pathological parameters of the cases are given in Table 1. Median age was 60 years. Ten (55.6%) patients were male and eight (44.4%) patients were female. Fifteen (83.3%) patients presented with stage IV disease. All CRC specimens, which included samples that metastasized to either the liver or lung only or metastasized to both the liver and lung, were obtained between June 2012 and April 2018, which was a longer time interval between sample collections and may increases the likelihood of detecting greater genomic differences. All these specimens were analyzed by the Pathology Center after obtaining appropriate human studies approval. Informed consent was obtained from every patient whose tissue sample was analyzed by targeted next-generation sequencing. For formalin-fixed and paraffin-embedded (FFPE) samples, tissues were processed and stained with hematoxylin and eosin (H&E) to confirm the presence of tumor cells (70% or greater).
Patient Characteristics.
Tissue DNA Extraction
DNA was extracted from FFPE tumor tissues using a QIAamp DNA FFPE tissue kit (Qiagen; CA, USA) according to the manufacturer's instructions. The nucleic acid concentration was determined using a NanoDrop1000 spectrophotometer (Thermo Fisher Scientific; Waltham, MA, USA).
Capture-Based Targeted DNA Sequencing
A minimum of 50 ng DNA was required for NGS library construction. Tissue DNA was sheared using a Covaris M220 focused-ultrasonicator, followed by end repair, phosphorylation, and adaptor ligation. DNA fragments with a size of 200-400 bp were selected by beads (Agencourt AMPure XP Kit), followed by hybridization with capture probe baits, hybrid selection with magnetic beads and polymerase chain reaction (PCR) amplification. An OncoScreen Plus panel consisting of 520 cancer-related genes spanning 1.6 Mb of the human genome was used. The quality and size of the fragments were assessed using a Qubit 2.0 fluorometer with a dsDNA high-sensitivity assay kit (Life Technologies, Carlsbad, CA). Indexed samples were sequenced on a NextSeq 500 sequencing system (Illumina, Inc., USA) with paired-end reads.
Sequence Data Analysis
Sequence data were mapped to a reference human genome (hg19) using Burrows-Wheeler Aligner 0.7.10 software. Local alignment optimization, variant calling and annotation were performed using Genome Analysis Tool Kit 3.2, MuTect, and VarScan software. Variants were filtered using the VarScan fpfilter pipeline, and loci with depths less than 100 were filtered out. Base calling in the tissue samples required at least 5 and 8 supporting reads for small insertion-deletions (INDELs) and single-nucleotide variants (SNVs), respectively. INDELs and SNVs with population frequencies greater than 0.1% in the ExAC, 1000 Genomes, dbSNP or ESP6500SI-V2 databases were grouped as single-nucleotide polymorphisms (SNPs) and excluded from further analysis. The remaining variants were annotated with ANNOVAR and SnpEff v3.6 software. Analysis of DNA translocation was performed using both Tophat2 and Factera 1.4.3 software.
Statistical Analysis
Data were summarized by frequency and percentage for categorical variables including mutation detection rate and distribution of mutation types. Comparisons between groups were performed using Fisher's exact test or chi-square test for these categorical variables wherever applicable. Wilcoxon signed-rank test or paired two-tailed Student's t-test was used for calculating the significance between continuous variables in two groups. All statistical tests were two-sided and differences were considered significant when P value was <.05. All data were analyzed using R statistics package version 3.4.0 (Vienna, Austria).
Results
Patient Characteristics
All patient characteristics are shown in Table 1. The median age at initial diagnosis was 60 years. We analyzed a total of 18 metastatic CRC cases, consisting of 47 tumors from 1 patient with liver and lung metastases (P1), 9 patients (P2-P10) with single liver metastases, and 8 patients (P11-P18) with single lung metastases. Two patients (P9 and P10) had liver metastases at the time of the initial surgeries, and the rest of the patients had no metastases at the time of the initial surgeries, but the metastases were found later. We used a novel and reliable method to detect microsatellite instability by NGS, 18 and all cases were classified as microsatellite stable (MSS) CRCs (Supplemental Table 1).
Mutational Profile of Colorectal Cancer Patients with Lung or Liver Metastases
A panel of 520 cancer-related genes spanning 1.6 Mb of the human genome was used. Generally, systemic mutations were detected in all samples, and most CRC-associated driver genes were also present at high frequencies (Figure 1A). Mutations in TP53 and APC were the most recurrent events and were observed in 78% and 72% of the samples, respectively. Other mutations, including KRAS, SMAD4 and ERBB3 mutations, which were always mutated in both primary and metastatic sites, were observed in 44%, 28% and 17% of the samples, respectively.

Mutation spectrum of patients in the cohort. (A) Mutation spectrum by individual sample. Patients labeled as P1, P2, etc. (B) Number of mutations identified in paired samples of CRC patients. (C) Mutation detection in CRC patients with liver and/or lung metastasis. Match represents shared mutations, Primary_specific represents mutations private to the primary tumor and Liver_specific and Lung_specific represent mutations private to the liver and lung metastases.
A total of 699 non-synonymous mutations were identified, 362 (51.8%) were shared between primary and matched tumors (Figure 1B and C; Supplemental Table 2), Interestingly, we found a higher correlation between primary and lung metastatic tumors than between primary and liver metastatic tumors (P = .021, Supplemental Table 3), indicating that liver metastases were more genetically different than lung metastases. Venn diagram analysis identified 72 specifically mutated genes in primary colorectal tumors, 51 in liver metastases and 24 in lung metastases. (Supplemental Figure 1 and Table 4), indicating a high degree of ITH in individual patients. In addition, the number of specifically mutated genes in primary tumors from patients with liver metastases was significantly greater than that in primary tumors from patients with lung metastases (P = .001), which suggests the likely polyclonal origin of primary CRC tumors that metastasize to the liver (P = .001, Supplemental Table 3). We also found that the TMB of the metastases was generally greater than that of the primary lesions. The TMB of the primary lesions of patients with liver metastases was greater than that of the primary lesions of patients with lung metastases, and the TMB of liver metastases was greater than that of lung metastases (P = .085, Supplemental Figure 2).
Differences in Functional Disruption in Multiple Pathways Between Primary Tumors and Metastases
Primary and liver metastatic tumor-specific mutations were consistently highly enriched for the PI3K-Akt, ErbB, Ras, and Rap1 signaling pathways (Figure 2A), suggesting a close relationship between primary and metastatic tumors. Compared with the matched primary tumor, lung metastases had a highly interconnected network of Rap1 and PI3K-Akt signaling pathways (P = .029, P = .029) (Figure 2B). Although there was no difference between liver and lung metastases (Figure 2C), there was a significant difference in their respective primary tumors (Figure 2D), especially in the estrogen signaling pathway, which targets cell cycle regulators, pro-apoptotic proteins, and cell adhesion molecules.
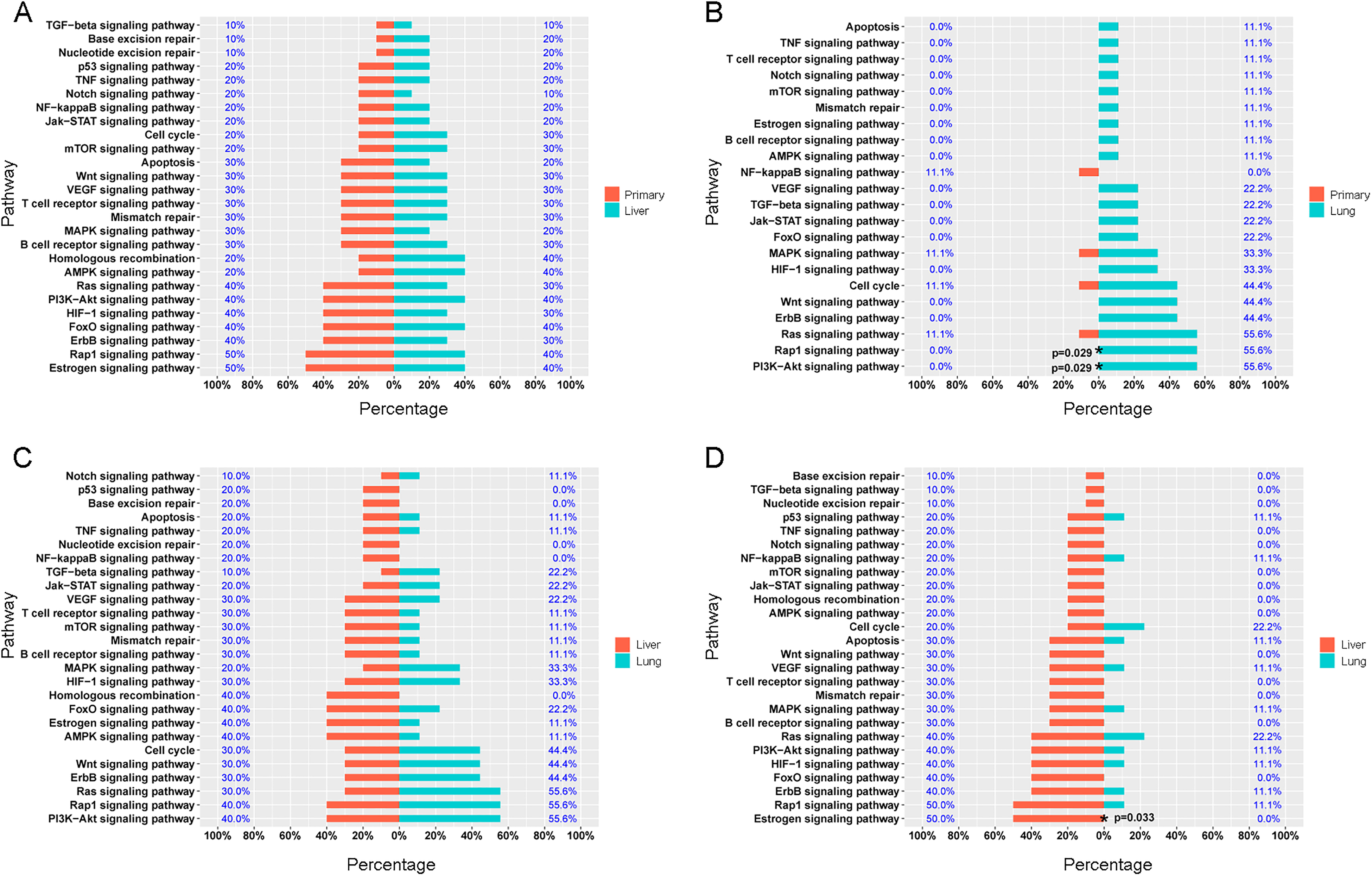
KEGG pathway analysis illustrating pathways found in (A) primary lesion and liver metastasis. (B) Primary lesion and lung metastasis. (C) Liver metastasis and lung metastasis. (D) Primary lesion in patients with liver metastases and lung metastases.
Phylogenetic and Clonal Evolution Analyses
For all CRC patients with multiple samples (≥3), we analyzed the regional distribution of mutations in P8, P9, and P10 (metastasis to the liver) and P11, P16, P17, and P18 (metastasis to the lung). We constructed a mutation spectrum and phylogenetic trees to depict the evolution and ordering of events.
For P8 (Figure 3A), liver metastases occurred at 1 and 3 years after diagnosing the primary tumors. The phylogenetic trunk represents ubiquitous mutations, including KRAS and TP53 mutations and DNMT3B, SRC, and TOP1 amplification. Furthermore, two liver metastases shared some genetic mutations accompanied by their own separate mutations, and there was a substantial genetic divergence between the primary and metastatic lesions. These findings suggest that liver metastases that disseminate from the primary tumor early show a substantial degree of genetic divergence, and the model of evolution was parallel progression. The same evolutionary model was observed in P10 (Figure 3C, Supplemental Figure 3), P11, P16, and P17 (Figure 4A-C). For P9 and P18 (Figures 3B and 4D), the metastases were seeded by the most advanced primary clones or subclones, and the degree of primary and metastatic genetic divergence was expected to be small; therefore, the metastases were more likely to have a linear progression model of evolution.

Regional distributions of somatic mutations and phylogenetic trees of three CRC patients with liver metastasis. (A-C) Heat maps showing the regional distribution of all mutations among primary and liver metastases in P8 and P9. Phylogenetic relationships in paired primary tumors and metastases of P8, P9, and P10. The blue line represents shared mutations in primary and metastasis tumors, the green line represents specific mutations in metastasis tumors, the red line represents shared mutations in metastasis tumors. Key mutations are marked with arrows. The lengths of the trunk and the branches of the tree are proportional to the number of corresponding mutations. P and M stand for the primary and metastasis tumor, respectively.

Regional distributions of somatic mutations and phylogenetic trees of four CRC patients with lung metastasis. (A-D) Heat maps and phylogenetic trees show the regional distribution and phylogenetic relationships in paired primary tumors and metastases of P11, P16, P17, and P18. The blue line represents shared mutations in primary and metastasis tumors, the green line represents specific mutations in metastasis tumors. Key mutations are marked with arrows. The lengths of the trunk and the branches of the tree are proportional to the number of corresponding mutations. P and M stand for the primary and metastasis tumor, respectively.
Clinical Relevance of Primary and Metastasis-Specific Mutations
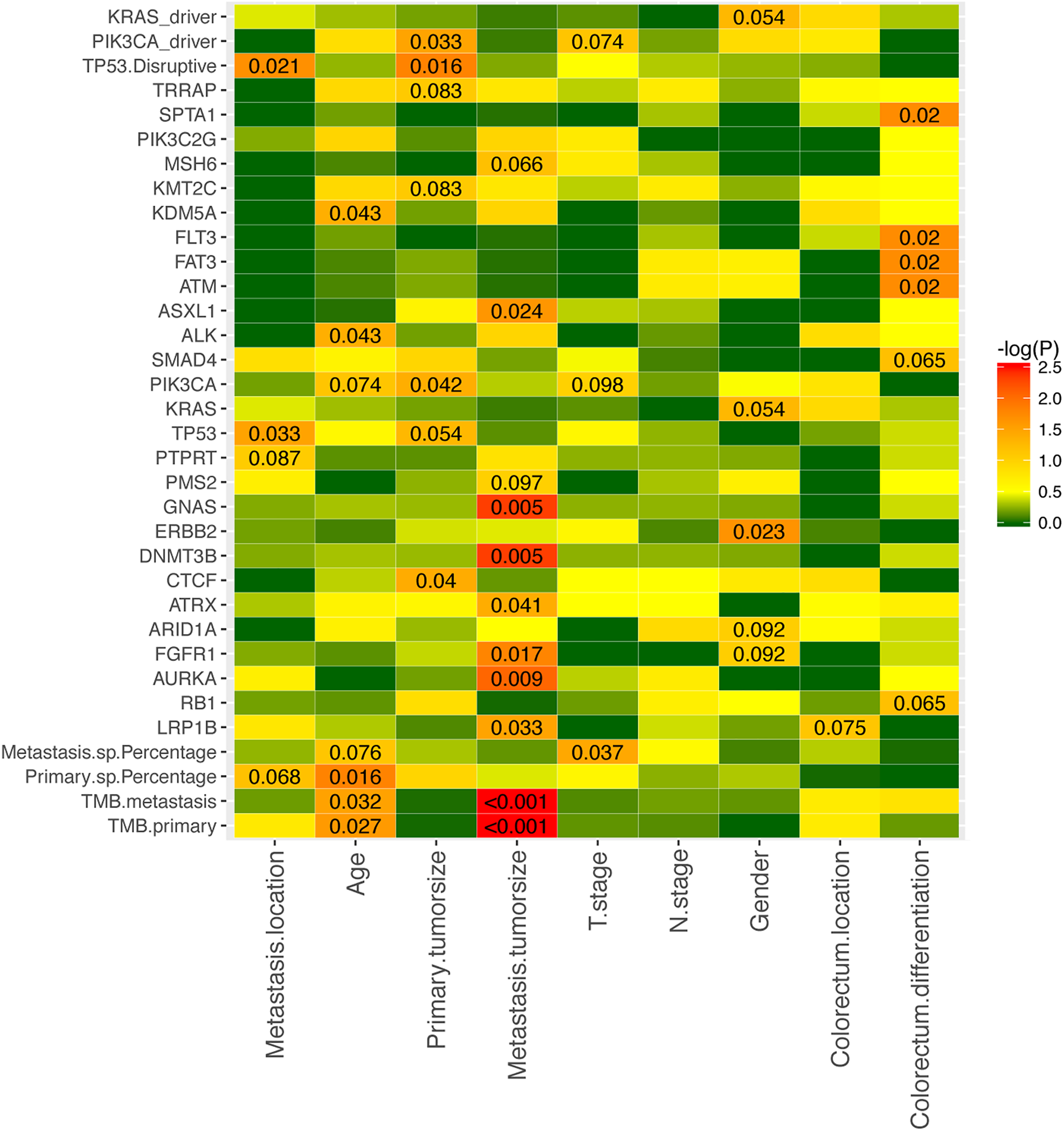
The 10 genes with the highest detection frequency of mutations in CRC with either liver or lung metastases included TP53, APC, LRP1B, PTPRT, FLT3, ASXL1, DNMT3B, FBXW7, FGFR1, and AURKA in CRC with liver metastases, TP53, KRAS, APC, SMAD4, SOX9, ERBB2, PIK3CA, PIK32G, ARID1A, and RB1 in CRC with lung metastases. As shown in Figure 5A-C, there was no significant difference in the mutation detected frequency of CRC patients between primary and metastatic tumor sites, but it was worth noting that AURKA and RB1 were mutated in only some of the liver and lung metastases, which are a characteristic mutation of metastases. Compared with patients with wild-type TP53, mutated TP53(P = .033), especially TP53-disruptive mutations (P = .021), indicated that tumor cells were more likely to metastasize to the liver. In contrast, tumor cells with PTPRT mutations were more likely to metastasize to the lung (P = .087) (Figure 6).

Comparison of mutation detection rate of various genes between the primary lesion and metastatic site in (A) all patients, (B) patients with liver metastasis, (C) patients with lung metastasis.

Percentage of CRC patients with TP53 and PTPRT mutation. (A-B) CRC patients with disruptive mutations in TP53 were more likely to have liver metastasis. (C) PTPRT-mutant CRC patients were more likely to have lung metastasis.
Notably, CTCF, PIK3CA, and TP53-disruptive mutations were significantly positively correlated with the size of the primary tumor (Figure 7, Supplemental Figure 4). Moreover, patients with AURKA, LRP1B, FGFR1, GNAS, DNMT3B, ATRX, or ASXL1 mutations had larger metastatic tumor sizes (Figure 7, Supplemental Figure 5). Notably, AURKA and LRP1B mutations were unique mutations in liver metastases and were significantly associated with metastatic tumor size (P = .009 and P = .033, respectively). In P7 and P10, both of whom had AURKA and LRP1B mutations, the size of liver metastases was significantly greater than that in patients with a single mutation. In addition, higher somatic TMB was associated with bigger metastasis tumor size (P < .001).
Clinical relevance of CRC patients with specific mutations.
Discussion
Our analyses showed that the coincidence mutation rate of primary tumors and metastases in patients with liver metastases was significantly less than that of lung metastases and established that more specific mutations in primary tumors from patients with liver metastases than in primary tumors from patients with lung metastases. This genetic heterogeneity has also been observed for metastases in CRC. 19 We hypothesized that CRC cells metastasized to the liver are more likely to overcome barriers to colonizing a metastasis site, which may depend on the subclones acquiring specific adaptive changes. Thus, liver remains a major metastasis-targeting organ of CRC. In addition, the prognostic value of the site of metastases has been shown in pancreatic adenocarcinoma 20 and breast cancers, 21 that is, lung metastasis indicates a better prognosis than liver metastasis. Furthermore, liver metastases have been shown to be more heterogeneous and not effective prognostic markers.
High TMB (52.2/Mb), which is defined as the top 20% of normalized mutational burden, has demonstrated benefits for immune checkpoint inhibitor (ICI) and overall survival in CRC patients. 22 Our study revealed that the TMB of primary and metastatic lesions in patients with liver metastases was greater than that in patients with lung metastases. The TMB in P10 was significantly greater than 52.2/Mb and exhibited not only the highest number of mutations detected in this study but also a distinct histologic subtype (Supplemental Figure 6). We concluded that a combination of morphological and genetic testing is more conducive to refinement of personalized immunotherapy.
Genetic alterations in the WNT, RAS-MAPK, PI3 K, TGF-β, and p53 pathways are common in CRC. 3 The PI3 K/AKT/mTOR pathway is a crucial and intensively explored intracellular signaling pathway in tumorigenesis.23–25 We found that the PI3K-AKT signaling genes were significantly mutated in lung metastases compared with paired primary tumors, suggesting that simultaneous inhibition of the PI3 K pathway may be required to achieve therapeutic benefits after CRC metastasizes to the lung. Our data also suggested that the incidence of PI3KCA and TP53 mutations was greater than that reported in previous studies on CRC.3,6,7,26,27 Considering the PI3KCA and TP53 mutation status, conflicting results have linked mutations to treatment responses and outcomes in CRC patients.27–31 In this study, we reported that CRC patients with disruptive mutations in TP53 were more likely to have liver metastases and that patients with PI3KCA mutations or TP53-disruptive mutations had larger primary tumor sizes.
The unique mutant genes that were only observed in liver and lung metastases were AURKA, CIC, CTNNB1, LATS2, PARP4, RAD52, TNFSF11, and CREBBP in liver metastases and FGFR1, HNF1B, KAT6A, KIT, MET, NF1, PIK3R1, SPEN, and TOP1 in lung metastases. Notably, AURKA was mutated in three out of nine CRC liver metastasis patients who had larger metastatic tumors. AURKA is also known to interact with mitotic regulators, which promote colorectal adenoma to carcinoma, 32 located on chromosome 20q. 33 Our findings further suggest that AURKA is a novel therapeutic target in liver metastases.
There are two general models of metastatic dissemination, the linear and the parallel progression models.34,35 P1 was the only patient whose liver and lungs were analyzed. The primary tumor and liver metastases were tightly clustered together, indicating their high genetic similarity (Supplemental Figure 7). The lung metastasis, which was resected 1.5 years after the resection of the primary tumor, had a drastically different genetic profile from that of the liver metastasis, suggesting that genetically distinct CRC cells from the primary sites might have migrated to the lung and formed the metastasis, likely independent of liver metastasis. These findings were also supported by the clustering analysis. As shown in Supplemental Table 5, analysis of P1 supported that the subclones from primary sites disseminated in parallel to both the liver and lung metastatic sites. The evolution model of multiple metastases formed in other patients was similar to that in P1. This model suggested parallel metastatic dissemination of tumor cells to distant organs independent of each other, and the few overlapping mutations indicated that the different lesion sites of the primary tumors might undergo independent mutation clonal expansions and become transformed as separate clones. These findings confirmed that genetic alterations that occur early in colorectal carcinogenesis persist throughout tumor evolution.
Previous studies have addressed that subclones of lymph node and distant metastases within the same CRC patient may have independent origins through 20-43 indels in polyguanine repeats by polymerase chain reaction (PCR) 36 and RNA sequencing was used to infer copy number variants (CNVs) combining targeted cDNA Sanger sequencing. 15 Moreover, the data revealed extensive intratumor heterogeneity both within tumors and between P/M pairs by analyzing exome sequencing data from 23 CRC patients with metastases to the liver or brain, and suggested that distant metastases are often seeded by a single clone (a single cell or a group of genetically similar cells). 37 To our study, the phylogenetic tree was built on the somatic mutations of 520 cancer-associated genes by next-generation sequencing, also indicates that a high degree of intertumor heterogeneity in individual tumors and distant lung or liver metastases arose from independent subclones in the primary tumor. Additionally, the study demonstrated that later dissemination results in more clonal mutations in the metastasis, many of which are at low frequency in the primary tumor and often undetectable in bulk sequencing. Then it gives rise to more metastasis-private clonal mutations in real sequencing data, leading to higher primary tumor-metastasis genomic divergence (PMGD). 37 Our data identified 72 specifically mutated genes in primary colorectal tumors, 51 in liver metastases, 24 in lung metastases, and CRC patients with liver metastases had more somatic mutations than patients with lung metastases in their primary tumors. This finding suggests that there are more potential functional mutations in liver metastases than lung metastases which are coherent with the high incidence rate of liver metastases than lung metastases in CRCs.
However, our research has several limitations. First, the main results of this study are based on a cohort of nine patients with CRC-liver metastasis and eight patients with CRC-lung metastasis. Although all patients in this cohort are MSS, the cohort size is small. Any results should be carefully interpreted, and more patients should be recruited to strengthen the conclusions. Second, to recall the clonal evolution from primary tumors and metastases, the subclones within primary and metastatic tumors are identified to reconstruct the tumor evolution process. The identification of reliable subclones more relies on whole exome sequencing and whole genome sequencing. Our data was built on the somatic mutations of 520 cancer-associated genes which was a small proportion of genomes. Therefore, our data may be not widely applicable and will need further corroboration in a larger cohort, but our data still highlighted the potential use of specific somatic mutations to survey tumor metastases, which could yield novel therapies.
Conclusions
Overall, our study showed that the genome landscapes of primary tumors and different metastases, are similar but certainly not identical especially between primary tumors and liver metastases, which supports the hypothesis of divergent evolution of metastatic lesions compared to primary tumors after truncal separation. So, investigating targeted therapies for metastases should ideally be based on the genetic properties of the metastases rather than on the genetic properties of primary tumors. Further analysis in the clinic is of utmost importance.
Supplemental Material
sj-docx-1-tct-10.1177_15330338231185285 - Supplemental material for Comprehensive Mutation Profiling of Colorectal Cancer Patients With Lung or Liver Metastasis by Targeted Next-Generation Sequencing
Supplemental material, sj-docx-1-tct-10.1177_15330338231185285 for Comprehensive Mutation Profiling of Colorectal Cancer Patients With Lung or Liver Metastasis by Targeted Next-Generation Sequencing by Chun-Ting Hu, Jing-Long Wang, Ting Hou, Zhao-Wen Yan, Li-Dong Zu, Guo-Hui Fu and Wei-Wei Shen in Technology in Cancer Research & Treatment
Footnotes
Abbreviations
Acknowledgments
We gratefully acknowledge the patients and family members who gave their consent on presenting the data in this study.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received the following financial support for the research, authorship, and/or publication of this article: This work was supported by the Shanghai Municipal Key Clinical Specialty (shslczdzk01303), National Natural Science Foundation of China (NO81972326, NO81372637, NO81602096), First Round of 3-year Action Plan to promote clinical skills and clinical innovation in Municipal Hospitals of Shanghai (16CR2039B).
Ethical Approval
The study was approved by the Ethics Committee of Shanghai General Hospital, Shanghai Jiao Tong University School of Medicine (no. 2019-KY020).
Supplemental Material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.