
Abstract
Keywords
Background
Colorectal cancer (CRC) is the third-most frequently diagnosed cancer and the second-largest cause of cancer-related fatalities worldwide. In 2020, more than 1.9 million patients with CRC were diagnosed globally, with an expected 935,000 deaths. 1 Approximately 20% of patients with CRC present with metastatic disease at the time of diagnosis of the primary tumor, and another 40% will develop metastases as the disease progresses.2,3 Patient prognosis for metastatic CRC (mCRC) is poor, with a 5-year survival rate of <15%. 4
Systemic therapy is the most promising approach to prolong survival and improve the quality of life of patients with unresectable mCRC. 3 Except for a small proportion of patients with mCRC (4%) with microsatellite instability-high (MSI-H) or mismatch repair-deficient (dMMR) status who can receive immune therapy, 5 first-line standard systemic treatments for most patients with mCRC involve fluorouracil, oxaliplatin, and irinotecan-based chemotherapies combined with antivascular endothelial growth factor (VEGF) or anti-epidermal growth factor receptor (EGFR) monoclonal antibodies such as bevacizumab, panitumumab, and cetuximab. The application of the anti-EGFR antibodies cetuximab and panitumumab is limited to left-sided and RAS/RAF wild-type (WT) mCRC, while anti-VEGF antibody is widely used in mCRC. 6 A series of clinical studies have shown that the addition of the anti-VEGF antibody bevacizumab, prolonged progression-free survival (PFS) and overall survival (OS) in patients with mCRC in comparison with those who received chemotherapy alone.7–11 In the subsequent trials to compare the effectiveness of anti-VEGF and anti-EGFR antibodies combined with chemotherapy, FOLFIRI or FOLFOX plus bevacizumab or cetuximab regimens showed no difference in PFS for patients with RAS WT mCRC.12,13 On the other hand, the results from 2 other trials showed that combination therapy with more than 1 biological agent, such as bevacizumab and cetuximab or panitumumab, is not associated with improved outcomes and can cause increased toxicity.14,15 Over the past 15 years, no new biological therapies have been approved as initial therapy for mCRC.
Rationale for Antiangiogenesis
On the basis of its role in the pathogenesis of CRC, angiogenesis has been proven to be a clinically meaningful target for the treatment of mCRC, and bevacizumab, an antiangiogenic monoclonal antibody, is widely used in mCRC treatment. Antiangiogenic tyrosine kinase inhibitors (TKIs) are small-molecule inhibitors that can stop the intracellular signaling system usually initiated when the receptor and ligand interact, suggesting that TKIs are a promising new treatment option. Antiangiogenic TKIs have been approved for multiple cancer types, including chronic myeloid leukemia, renal cell carcinoma (RCC), nonsmall cell lung cancer (NSCLC), and CRC, and have shown high selectivity, high efficacy, low side effects, and ease of preparation. 16 TKIs, including regorafenib and fruquintinib, have been approved as third-line therapy for mCRC,17–19 but previous efforts to develop combination therapies of TKIs with chemotherapy as the first-line treatment for mCRC have been unsuccessful.20–23 Nevertheless, these studies have underscored the importance of exploring further treatment options for mCRC to address the key unmet needs.
Anlotinib is an oral small-molecule TKI that mainly targets anti-VEGF receptors 1/2/3, fibroblast growth factor receptors 1-4, and platelet-derived growth factor receptors α/β.24,25 It has been approved for the treatment of patients with NSCLC, small cell lung cancer, soft tissue sarcoma, medullary thyroid cancer, and radioiodine-refractory differentiated thyroid cancer in China.26–30 Furthermore, anlotinib has been confirmed to show anticancer activity in several other advanced malignant cancers, including esophageal squamous cell cancer, hepatocellular carcinoma, RCC, and CRC.31–34 A phase III trial, ALTER-0703, demonstrated the efficacy and tolerability of anlotinib monotherapy in patients with refractory mCRC. 34 This study showed that anlotinib significantly prolonged PFS in comparison with placebo (4.1 vs 1.5 months; hazard ratio [HR] = 0.34; P < .0001), although OS did not differ significantly between the anlotinib and placebo groups (8.6 vs 7.2 months; HR = 1.02; P = .870). Subgroup analysis demonstrated an OS benefit in patients with KRAS/NRAS/BRAF WT mCRC (HR = 0.68, 0.47-0.99), indicating that these patients are potential candidates for anlotinib therapy.
The common adverse reactions associated with anlotinib include hypertension, fatigue, hand-foot syndrome, hypertriglyceridemia, proteinuria, diarrhea, decreased appetite, increased blood thyrotropin levels, hypercholesterolemia, and hypothyroidism. These adverse events can be controlled or relieved by dose reduction or symptomatic treatment, respectively. The incidence of grades 3 or 4 bleeding events was approximately 1.6%, while that of grade 5 was 0.4%, based on the findings for 1788 patients with advanced tumors in 22 clinical trials.
We had previously conducted a phase II study of anlotinib in combination with oxaliplatin and capecitabine as a first-line therapy for patients with RAS/BRAF WT metastatic colorectal adenocarcinoma (ALTER-C-002). The objective response rate (ORR) and disease control rate (DCR) were 73.3% and 93.3%, respectively. The median PFS was 11.3 months, which demonstrated the preliminary effectiveness of anlotinib combined with CAPEOX for mCRC. 35 The safety profile was tolerable, and no unexpected toxicity was identified. Therefore, we launched this multicenter, randomized, parallel-controlled, phase III trial to further assess the efficacy and safety of anlotinib plus CAPEOX as a first-line treatment for patients with RAS/BRAF WT mCRC.
Methods
Study Objectives and Design
The objective of this phase III study is to compare the efficacy and safety of anlotinib plus CAPEOX versus bevacizumab plus CAPEOX as a first-line therapy for RAS/BRAF WT mCRC.
This is a multicenter, randomized, open-label, parallel-controlled, non-inferiority, phase III study. Figure 1 presents the study design. The study was initiated in May 2021 and will be conducted at approximately 70 high-quality sites in China. The World Health Organization Trial Registration Dataset is attached as a supplementary file.

Flow diagram of the study design.
Eligibility Criteria
A total of 698 patients with histologically or cytologically confirmed colorectal adenocarcinoma and unresectable metastases assessed by a multidisciplinary team (MDT) will be enrolled. Eligible patients, aged 18 to 75 years, must have an Eastern Cooperative Oncology Group) performance status of 0 or 1, with confirmed KRAS, NRAS, and BRAF WT, and at least 1 measurable lesion according to the Response Evaluation Criteria in Solid Tumors (RECIST) version 1.1. 36 Eligible patients should have tumor tissue genotyped for KRAS, NRAS, and BRAF mutations individually or as part of a next-generation sequencing panel.
Patients will be excluded if they are pregnant or lactating or have MDT-assessed potentially resectable metastases, MSI-H or dMMR, any comorbidity or conditions that can prevent tolerable treatment such as inadequate bone marrow, renal, or liver function; previous intolerance to capecitabine or oxaliplatin; recent serious cardiovascular events; active bleeding; or arteriovenous thrombotic events. Patients who have previously received systemic therapy for mCRC, those who received oxaliplatin-based adjuvant therapy within 12 months before enrollment, and those who received adjuvant therapy without oxaliplatin within 6 months before enrollment will also be excluded. Patients who had a history of treatment with antiangiogenic or anti-EGFR-targeted agents (including, but not limited to, bevacizumab, cetuximab, panitumumab, or aflibercept) or had contraindications for chemotherapy will also be excluded. Detailed inclusion and exclusion criteria are listed in Table 1.
Patient Eligibility Criteria.

Abbreviations: NMPA, National Medical Products Administration; ULN*, upper limit of normal; CTCAE, Common Terminology Criteria for Adverse Events.
Randomization and Stratification
The patients will be randomized in a 1:1 ratio using an eRand Central Randomization System. The enrolled patients will be stratified by primary tumor location (right vs left) and treatment history of adjuvant chemotherapy (yes vs no). Investigators will generate the allocation sequence, enroll participants, and assign them to the interventions.
Treatment Schedule
Interventions and Procedures
Figures 1 and 2 present the enrollment and intervention schedules for the experimental and control groups, respectively. The experimental group will receive CAPEOX in combination with anlotinib and the control group will be treated with CAPEOX plus bevacizumab. Patients in both groups will receive 4 to 8 cycles of induction treatment (CAPEOX plus anlotinib or bevacizumab) according to their tolerance, followed by maintenance treatment (capecitabine plus anlotinib or bevacizumab) until radiological or clinical progression, patient withdrawal, unacceptable toxicity, or the investigator's decision. Patients who are treated with fewer than 4 cycles of induction treatment due to objective factors can also be treated with the maintenance regimen and undergo follow up. Clinical research associates will regularly monitor the usage and records of drugs to guarantee adherence to the intervention protocols.
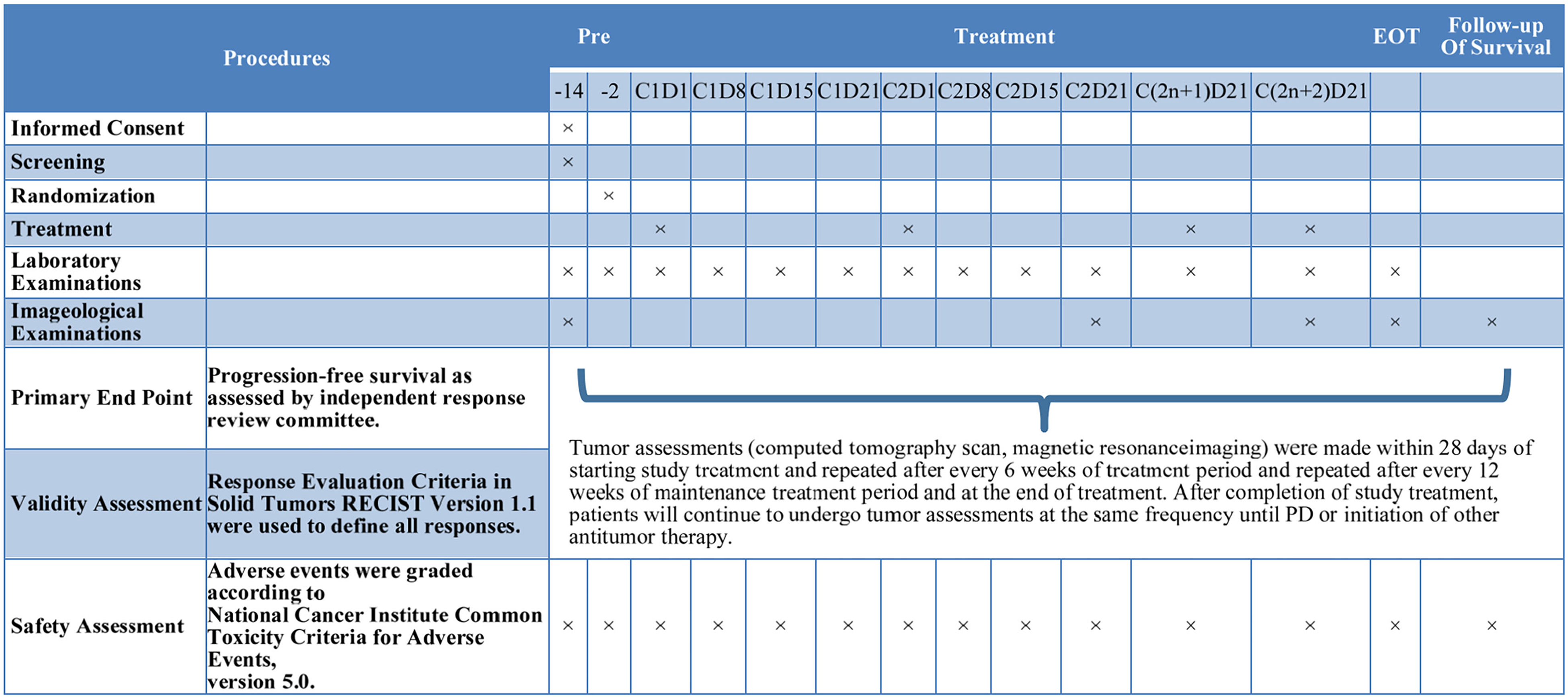
Schedule of treatment.
Dosing Schedule of the Investigational Drugs
For the induction treatment, patients randomized to the experimental group will receive anlotinib (12 mg p.o. qd, on days 1-14 every 21 days), oxaliplatin (130 mg/m2 ivgtt., on day 1 every 21 days), and capecitabine (850 mg/m2 p.o. bid, on days 1-14 every 21 days). Patients randomized to the control group will receive bevacizumab (7.5 mg/kg ivgtt., on day 1 every 21 days), oxaliplatin (130 mg/m² ivgtt., on day 1 every 21 days), and capecitabine (1000 mg/m2 p.o. bid, on days 1-14 every 21 days). For maintenance treatment, the patients in the experimental group will receive anlotinib (12 mg p.o. qd, on days 1-14 every 21 days) and capecitabine (850 mg/m2 p.o. bid, on days 1-14 every 21 days). Patients in the control group will receive bevacizumab (7.5 mg/kg ivgtt., on day 1 every 21 days) and capecitabine (1000 mg/m2 p.o. bid, on days 1-14 every 21 days).
Concomitant Medications
No anticancer agents, including modern Chinese medicine preparations and immune modulators, other than the study medications, will be administered to the patients. The use of bisphosphonates for bone metastases, palliative treatment for nontarget lesions, and radical surgery for patients whose tumors regress to the surgical resection criteria during the study period will be allowed.
Endpoints
The primary endpoint is PFS assessed by an independent review committee (IRC). The secondary endpoints are investigator-assessed PFS, OS, ORR, DCR, duration of response (DOR), resection rate of liver metastases (LMs), quality of life (QoL), and safety.
PFS is defined as the time from randomization to the first documented disease progression or death from any cause, whichever occurred first. OS is defined as the time from randomization to death from any cause. ORR will be calculated as the sum of the proportions of cases showing complete response (CR) and partial response (PR), while DCR will be calculated as the sum of the proportions of cases showing CR, PR, and stable disease. DOR is defined as the time from the first response to disease progression or death from any cause. The resection rate of LMs is the proportion of patients who undergo hepatectomy for curative intent after treatment. QoL will be assessed by the patient-completed European Organization for Research and Treatment of Cancer Quality of Life Questionnaire (EORTC QLQ-C30) 37 and EuroQoL 5-dimensional instrument (EQ-5D) questionnaire. 38
Assessment Schedule and Follow Up
In both groups, tumor assessments will be performed using cervical, thoracic, and abdominal enhanced computed tomography or magnetic resonance imaging (according to RECIST version 1.1). Tumor marker assessment will be performed at baseline and every 6 weeks during the induction period. For patients receiving maintenance therapy, tumor assessment will be performed every 12 weeks until radiological progression or the end of the study. Laboratory examinations, including complete blood count tests, blood biochemistry assessments, and electrocardiogram examinations, will be performed at baseline, every 7 days during the first 2 cycles, and every cycle thereafter. QoL will be assessed at baseline, every 2 cycles, and at the withdrawal visit. Standard safety monitoring will be performed from the time the patients provide informed consent to 30 days after the last dose, and adverse events (AEs) will be graded according to the Common Terminology Criteria for Adverse Events (CTCAE) version 5.0 (Figure 2). Tumor assessments will be performed during follow up for patients who have discontinued treatment without documented progressive disease. After disease progression, patients will undergo follow-up visits for documentation of survival status and post-study anticancer treatment and responses.
Statistical Considerations
Required Number of Patients
The sample size calculation of the non-inferiority phase III trial was based on a clinical study (Bevacizumab in Combination With Oxaliplatin-Based Chemotherapy As First-Line Therapy in Metastatic Colorectal Cancer: A Randomized Phase III Study). 7 We calculated a sample size of 349 patients per group, assuming a type I error of 0.025 (one-sided), a true HR of 0.85, a non-inferiority margin of 1.09 in terms of HR, and a 1:1 ratio of the sample sizes for the 2 groups. The power of the study to indicate non-inferiority was approximately 81.2%. Non-inferiority was established if the upper limit of the 95% confidence interval (CI) for HR was <1.09.
Finally, we calculated that the number of PFS events for the primary analysis was 524, and the total sample size was 698 cases, with 349 cases in the experimental group and 349 cases in the control group.
Recruitment
Principal investigators at more than 70 sites will review the diagnosis in the medical records to identify potentially eligible patients. Patients who agree to participate will receive an informed consent form (ICF) containing a full description of the study.
Data Management
The sponsor is responsible for data management using an electronic data collection system (EDC). The EDC will automatically record the inspection marks of all role operations such as data preservation, modification, deletion, proofreading, review, freezing, electronic signature, and lock.
Statistical Analysis Plan
The intention-to-treat (ITT) set includes all randomized patients based on the ITT principle. The per-protocol set (PPS) includes patients who completed at least 2 cycles of treatment and 1 efficacy assessment after randomization without major protocol deviations. The safety analysis set (SAS) includes all randomized patients who have taken at least 1 dose of the study medication. Assessment of baseline characteristics and non-inferiority analyses will be conducted in the ITT set. Efficacy will be assessed in the ITT set and the PPS. Safety will be assessed in the SAS. Maximum likelihood estimation would be adopted to handle the missing data.
Based on the Cox proportional hazards model, we will estimate the HR and 95% CI in the ITT and per-protocol populations with a one-sided type I error of 0.025. Kaplan–Meier analyses will be used to estimate the median PFS, OS, and DOR. The log-rank test will be used to assess differences between the 2 groups.
For PFS assessed by IRC, if the upper limit of 95% CI in terms of HR is less than the non-inferiority margin of 1.09, the efficacy of anlotinib combined with CAPEOX chemotherapy as first-line treatment for mCRC will be considered to be non-inferior to that of bevacizumab combined with CAPEOX chemotherapy. For ORR, DCR, and the resection rate of LMs, Chi-square test or Fisher's exact test will be used for comparisons between groups, and the Clopper–Pearson method will be used to calculate 95% CI.
The primary and secondary endpoints will also be compared between the treatment arms using a stratified log-rank test, with stratification factors as entered in the eRand Central Randomization System. The EQ-5D will be evaluated based on descriptive statistical analysis, calculating the number and percentage of different levels of each dimension in the 2 groups, and using the Wilcoxon rank-sum test for comparisons between groups. The EORTC QLQ-C30 will also be evaluated based on descriptive statistical analysis after calculating standardized scores, and the Wilcoxon rank-sum test will be conducted for comparison.
Based on descriptive statistical analysis, the AEs that occur in the 2 groups in this trial will be described in a list, and Fisher's exact probability method will be used to compare the incidence of AEs, if necessary.
Confidentiality
Measures to protect confidentiality can be summarized as follows: only a unique study number will identify patients in the electronic case report form (eCRF) or other documents to be submitted to the sponsor. Patient names and other unnecessary personal information will not be entered in the eCRF. No material bearing a patient's name will be kept on file by the sponsor. The patients will be informed of their rights within the ICF. The principal investigator and sponsor will have access to the final trial data set and no media publicity will be allowed during the study period.
Discussion
The results of this randomized, phase III study that prospectively compares the antitumor outcomes of standard first-line systemic therapy (CAPEOX plus bevacizumab) and CAPEOX with anlotinib in patients with RAS/BRAF WT unresectable mCRC will reveal whether the oral TKI anlotinib combined with CAPEOX can produce non-inferior or even better clinical outcomes for patients with RAS/BRAF WT mCRC.
For unresectable mCRC, anti-VEGF or anti-EGFR antibodies plus chemotherapy are the standard first-line treatment, depending on the RAS/BRAF status. Both anti-VEGF and anti-EGFR monoclonal antibodies are available for WT RAS/BRAF. The median PFS of chemotherapy alone for mCRC is 8 months, while the addition of bevacizumab increased the mPFS to approximately 10 months.7,8
In the GALGB 80405 and FIRE-3 clinical trials, patients with unresectable KRAS WT mCRC were treated with bevacizumab or cetuximab plus chemotherapy as first-line treatment: the median PFS was 10-10.5 months in the cetuximab-chemotherapy group and 10.3-10.6 months in the bevacizumab-chemotherapy group, and the response rates were 59.6% to 62% versus 55.2% to 58% for cetuximab and bevacizumab, respectively, which were not significantly different.12,13 On the other hand, 2 phase II clinical trials (ALTER-C-001 and ALTER-C-002) evaluated the efficacy and tolerance of anlotinib with CAPEOX as a first-line treatment in unresectable mCRC. In ALTER-C-001, the ORR was 62.5% and median PFS was 7.79 months regardless of the RAS and RAF status, 39 and in ALTER-C-002, the mPFS was 11.4 months and the ORR was 73.3% in RAS/RAF WT mCRC. These 2 trials indicate that anlotinib combined with CAPEOX offers potential clinical benefits in patients with unresectable mCRC. 35
In terms of the safety profile, the most common grade ≥ 3 treatment-related adverse events (TRAEs) of anlotinib monotherapy in mCRC were hypertension, increased γ-glutamyl transpeptidase (γ-GT), and hand-foot skin reaction. When combined with oxaliplatin and capecitabine, the most common grades 3 or 4 TRAEs (>10%) were hypertension, decreased neutrophil count, and diarrhea, which can be controlled by dose reduction or delay. The combination of anlotinib and CAPEOX is generally well tolerated, and there is no evidence to suggest that anlotinib increases the frequency or severity of the known toxicities of oxaliplatin and capecitabine.
Moreover, anlotinib is an oral small-molecule multitarget TKI, which offers a series of conveniences, especially during maintenance treatment; patients only receive anlotinib and capecitabine orally and do not need to stay in a treatment facility. We hope that oral TKIs could provide more choices for patients with unresectable mCRC with clinical benefits comparable to the present standard of care.
Thus, based on the previous phase II study data, it is very meaningful to launch a multicenter, randomized, phase III clinical trial to compare the efficacy and safety of the anlotinib plus CAPEOX regimen with the standard treatment for unresectable mCRC patients with RAS/BRAF WT.
Conclusion
This phase III study is designed to investigate the efficacy and safety of anlotinib combined with CAPEOX as a first-line therapy for patients with RAS/BRAF WT mCRC. The results of this trial will potentially lead to anlotinib being a new treatment option for mCRC, with improved survival and decreased toxicity.
Supplemental Material
sj-pdf-1-tct-10.1177_15330338231152350 - Supplemental material for A Randomized Phase III Study of Anlotinib Versus Bevacizumab in Combination With CAPEOX as First-Line Therapy for RAS/BRAF Wild-Type Metastatic Colorectal Cancer: A Clinical Trial Protocol
Supplemental material, sj-pdf-1-tct-10.1177_15330338231152350 for A Randomized Phase III Study of Anlotinib Versus Bevacizumab in Combination With CAPEOX as First-Line Therapy for RAS/BRAF Wild-Type Metastatic Colorectal Cancer: A Clinical Trial Protocol by Jinjie He, Yue Liu, Chengcheng Liu, Hanguang Hu, Lifeng Sun, Dong Xu, Jun Li, Junye Wang, Xiaobing Chen, Rongbo Lin, Yi Jiang, Yanqiao Zhang, Weisheng Zhang, Ying Cheng, Xiaohong Wu, Mingzhi Fang, Enxiao Li, Ye Xu, Ye Chen, Jiayi Li, Yanyan Cui, Zhanyu Pan, Songnan Zhang, Ying Yuan and Kefeng Ding in Technology in Cancer Research & Treatment
Footnotes
Acknowledgments
The authors thank the patients and their families as well as the investigators, co-investigators, the study teams at each of the participating centers and Chia Tai Tianqing Pharmaceutical Group Co., Ltd.
Availability of Data and Material
All data generated or analyzed during this study are included in this published article.
Ancillary and Post-Trial Care
Clinical research insurance was bought for all patients, and patients who suffer harm from trial participation will get compensation and post-trial care.
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: The clinical trial is sponsored by Chia Tai Tianqing Pharmaceutical Group Co., Ltd.
Authors’ Note
KFDing is responsible for trial conception and design, and manuscript revision. JJ He, Y Liu, and CC Liu were responsible for data collection and analysis, and drafting the manuscript. HG Hu, LF Sun, D Xu, J Li, JY Wang, XB Chen, RB Lin, Y Jiang, YQ Zhang, WS Zhang, Y Cheng, XH Wu, MZ Fang, EX Li, Y Xu, Y Chen, JY Li, YY Cui, ZY Pan, SN Zhang, and Y Yuan are responsible for data colletion and manuscript revision. All authors reviewed and approved the final version for submission.
The principal investigator is responsible for collection, and interpretation of data; and the decision to submit the report for publication and will permit study-related monitoring and inspections by the sponsor or its representative(s), government regulatory authorities, and the Institutional Review Board(s) of all study-related documents. Chia Tai Tianqing Pharmaceutical Group Co., Ltd will be in charge of data management. Department of Biostatistics, School of Public Health, Nanjing Medical University has a role in data analysis.
Ethical Approval and Consent to Participate
This clinical trial was approved by the National Medical Products Administration (NMPA) of China in accordance with the Declaration of Helsinki (2013 Edition) 40 and relevant Chinese clinical trial research norms and regulations. The study centers should be qualified for systemic treatment of patients with mCRC. Written informed consent will be obtained from all patients enrolled in the trial. The study protocol (Date March 15, 2022, Version 2.1) was approved by the Human Research Ethics Committee of the Second Affiliated Hospital of the Zhejiang University School of Medicine(approval number: (2022) LSYD No. (257), approval date: 12/4/2022, location: Hangzhou China). The study protocol was also approved by all other participant centers. The principal investigator or sponsor (as appropriate) must submit and, where necessary, obtain approval from the Institutional Review Board(s) and/or Independent Ethics Committee(s) at those centers for all subsequent protocol amendments and changes to the informed consent document. If required, the sponsor will also ensure that the implementation of substantial amendments to the protocol and other relevant study documents occurs only after approval by the relevant regulatory authorities. Central ethical approval has been obtained from the Human Ethics Committee of the Second Affiliated Hospital of the Zhejiang University School of Medicine. The trial has been registered at clinicaltrials.gov (registration date: April 2021, NCT04854668) with the title of “A study of anlotinib hydrochloride capsule combined with chemotherapy as first-line treatment in subjects with RAS/BRAF wild metastatic colorectal cancer.” All the patients enrolled in the trial will be required to provide written informed consent. The trial will not begin at a center until written approval has been obtained from the Institutional Review Board(s) and/or Independent Ethics Committee(s) at each center.
Supplemental Material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.