
Abstract
Introduction
The investigation of extracellular vesicles (EVs) has gained importance in recent years due to the significance and possible applications of these structures in biomedical sciences. The family of EVs is commonly considered to consist of apoptotic bodies, which are the largest EVs, endosomes, and exosomes. 1 When first reported, exosomes were seen as fragments of cellular vesicles or membranes; later, these structures were described as small vesicles which can act as cellular “garbage bags,” and, more recently, exosomes have been proposed to be specific cellular components tasked with orchestrating different aspects of specific immune responses. 2
Rose Johnston was the first to use the term “exosomes” and demonstrate the enzymatic activity of these structures. 3 Exosomes are the smallest of the EVs generated by cells, with sizes ranging between 30 and 150 nm. In terms of identification, exosomes are characterized chiefly by the presence of members of the tetraspanin family, such as CD63, CD9, CD81, ALIX, and HSP70.1,4
These microvesicles are generated in the endocytic pathway: the first step is endocytosis (which may be mediated by various mechanisms), then the endosome will fuse with several microvesicles (which originate in various membranous compartments such as the Golgi, nucleus, and endoplasmic reticulum), forming a multivesicular body (MVB), then the MVB has 2 possible paths: fuse with the lysosome, thus leading to the degradation of its contents, or fuse with the cell membrane, leading to the liberation of the contents of the MVBs to the extracellular space. It is here, in the later step, where exosomes are formed: once MVBs release these vesicles outside of the cell, these vesicles take on the name of exosomes. 5
Due to their complicated and varied biogenesis, the cargo of exosomes is likewise varied and vast, from DNA and RNA to lipids and a notable variety of proteins; today exosomes are considered to be one of the most novel and important mechanisms of cell–cell communication.4,6
Generally speaking, the contents of a given population of exosomes may vary depending on the cell that generated the exosomes. Thus, exosomes can play dual roles in the immune response; that is to say, exosomes can both activate and downregulate the activities of immune cells. Via their capacity to traffic molecules such as miRNAs, peptide-MHC I/II complexes, or diverse cytokines, exosomes have been shown to promote the activities of monocytes, macrophages, dendritic cells, B cells, T cells, and NK cells. 7
Likewise, exosomes have also been shown to modulate the immune response. Exosomes have been implicated in escape pathways for infectious agents such as bacteria, viruses, and transformed cells. In cancer, it has been reported that exosomes can induce transformation and promote immune escape, tumor growth, angiogenesis, metastasis, and drug resistance. These varied effects have been reported to be due to the activity of exosomes containing damaged or transformed DNA, miRNAs, enzymes such as metalloproteases, and immune ligands or receptors. Additionally, the presence of these molecules in exosomes, either as exosomal cargo or anchored to the external face of the exosomal membrane, has served to define unique populations of exosomes that may serve as tumor biomarkers or possible therapeutic targets.1,8,9
One notable example of the potential importance of exosomes in modulating the immune response is seen in the case of immune checkpoint ligands. The exploitation of immune checkpoint receptors and their ligands as therapeutic targets has exploded in recent years. Principal among these is the case of the inhibitory receptor PD-1 (found on a subset of activated antitumor cytotoxic T and NK cells) and its ligands PD-L1 and PD-L2 (found on tumor cells and other cells in the tumor microenvironment). The presence of different forms of exosomal PD-L1 in various types of cancer has been reported; these exosomes promote immune escape and tumor growth by decreasing the number and cytotoxicity of CD8 + T cells.10–12
Working with exosomes or other EVs is challenging due to their phenotypic characteristics such as their minimal size, their origin from very different cells, and presence in all tissues or body fluids. There are diverse techniques for isolating, measuring, and quantifying EVs; nevertheless, the gold standards are ultracentrifugation and transmission electron microscopy. Among other techniques for isolation are a wide variety of centrifugation-based techniques such as differential centrifugation or density gradient centrifugation, to mention a few. Capture-based techniques use agents such as beads covered by antibodies against classic biomarkers on the exosome membrane such as CD63. We can use precipitation agents such as polyethylene glycol or chromatography-based techniques to isolate the exosomes; however, the challenge of the purity of the sample remains. To measure their size and concentration, nanoparticle tracking analysis is one of the predominant techniques along with resistive pulse sensing. In the protein field, the most used techniques are Western blot, which is qualitative and semiquantitative, ELISA, and mass spectrometry for proteomic analyses.13–15
One of the most promising approaches in the study of EVs is flow cytometry. This method allows us to detect, measure, and identify the exosomes and their cargo molecules with the use of fluorescent antibodies; however, most cytometers are not able to measure particles as small as exosomes. Due to this, the use of capture beads is necessary for the cytometric analysis of exosomes, but this is often problematic, as the exosome sample will usually contain artifacts such as cellular debris, which may also bind to the capture beads. Because of this, background noise can be significant, and the fluorescent signals of the targets will be relatively dim. For these reasons, there is still no consensus about how to properly isolate and work with these EVs,2,13–15 especially when using flow cytometry. Here, we present an adapted technique for the preparation of exosomes and their analysis using conventional flow cytometry.
Materials and Methods
The complete protocol that we used is summarized in Figure 1. The details of the steps utilized are as follows:

A schematic summary of the protocol.
Sample Collection and Preparation
We utilized 10 samples from healthy donors and 10 samples from cervical cancer patients. Peripheral blood (14 mL) was collected by venipuncture using tubes without anticoagulants (BD Vacutainer ® Serum Tubes). The blood was allowed to stand for 30 min, and then the blood was centrifuged at 1500 rpm for 15 min at room temperature. The serum was then separated into tubes and stored at −80°.
This study was submitted to the relevant institutional review board committees (Comisiones de Investigación, Ética y Bioseguridad del Centro Universitario de Ciencias de la Salud, Universidad de Guadalajara) and was approved and classified as a study without undue risks or burdens that complied with the institutional requirements ensuring appropriate ethical and biosecurity conduct; this study was registered as CI-02117. All patients that participated signed letters of informed consent.
Isolation of Exosomes and Western Blot
Exosome isolation was performed with the Total Exosome Isolation Reagent (Cat. 4478360, Invitrogen) according to the manufacturer's instructions. After defrosting, samples were centrifuged at 2000 g/30 min to remove any cellular debris. The isolation reagent (100-150 μL) was then added to the debris-free sera (at a ratio of 100 μL reagent : 500 μL serum), which was subsequently vortexed and then incubated for 30 min at 2 °C to 8 °C. The samples were next centrifuged at 12 000 g/10 min and the exosome pellet was resuspended in PBS by vortexing. The amount of protein in each sample was determined by spectrophotometry with a NanoDrop One (ThermoFisher Scientific). The samples (denatured with DTT at 95 °C for 5 min, 80 µg/lane) were resolved and analyzed by SDS-PAGE and immunoblotting using the classical marker of vesicles with features of exosomes (anti-CD63 cat. sc-5275, lot number C1317, Santa Cruz Biotechnology). Primary antibody binding was detected using mIgG Kappa Binding Protein-HRP, sc-516102, lot C1418, Santa Cruz Biotechnology.
In order to ensure that only whole exosomes were binding to the beads, in a control group the exosome fraction was additionally treated with detergents, 1% SDS, Tween 20, Triton x-100, all from Sigma-Aldrich. The selected samples were incubated with the detergents for 5 min, then vortexed for 2 min, before proceeding to the bead coupling step.
Coupling of Beads With Exosomes
The exosomes were coupled to the magnetic beads using the Exosome – Human CD63 Isolation/Detection Reagent (from cell culture media) (Cat. 10606D, Invitrogen).
After exosome isolation, varying amounts of exosomes (from 10 to 200 µg) were mixed with varying volumes (6.67-40 μL) of magnetic beads. In the optimized protocol, 200 μg of exosomes were incubated with 20 μL (200,000 beads) of magnetic beads per tube. PBS was added to each tube to reach a final volume of 100 μL, and then the beads and exosomes were incubated for 18 to 22 h at 2 °C to 4 °C.
Staining for Flow Cytometry
On the next day, after overnight exosome-bead coupling, the beads were stained with antibodies. The beads were stained with differing antibodies depending on the experiment, but 5 μL of all antibodies and isotype controls were used. Anti-CD9 APC (Cat. 312108, Lot B338122, Biolegend), and anti-CD81 APC (Cat. 349510, Lot B340368 Biolegend) were used to identify exosome markers. Anti-PDL1 PE (Cat. 329706, Lot B3429175, Biolegend), and anti-CD155 PE-Cy7 (Cat. 337614, Lot B337215 Biolegend) were used to identify ligands of interest, and the following isotype controls were also used: APC (Cat. 400120, Lot 243040, Biolegend), PE (Cat. 400314, Lot B229091, Biolegend), and PE-Cy7 (Cat. 400126, Lot B308653 Biolegend). After the addition of the antibodies, the tubes were incubated at room temperature on a shaker for 1 h.
After the incubation, the tubes were centrifuged at 200 g for 10 min, and the unbound beads/antibodies/PBS was carefully aspirated with a pipette. Then 500 μL of PBS was added and pipetted up and down to wash the pelleted beads + exosomes.
The tubes were placed on the magnet (BD Imag; Becton Dickinson) for 1 min and the nonmagnetically bound fraction was decanted. Then the tubes were removed from the magnets and 500 μL of PBS was added to each tube prior to reading with the cytometer.
An Attune Acoustic Focusing Cytometer (ThermoFisher Scientific) was used. The analyses were performed with Kaluza Analysis software from Beckman Coulter and the graphics with GraphPad Prism 6 (GraphPad).
Results
After testing variable concentrations and wash steps, we found our highest exosome yield was when we used 200 µg of protein with 200,000 beads, a modified washing protocol, and a select region in the flow cytometry analysis. Unbound beads and multiple bead:exosome:bead complexes are significant complications to the use of this technique. We confirmed the identity of the bead:exosome population with the highest concentration of exosome markers. We also confirmed, using western blot for CD63, the presence of this exosome marker in varying concentrations of the exosomal fraction prior to bead coupling (Figure 2B). We confirmed the specificity of the exosome isolation by using a Western blot for CD63 and demonstrated the lack of nonspecific binding when the exosome-free fraction was incubated with anti-CD63; likewise, we demonstrated the binding specificity of the PD-L1 antibody, which we used in the flow cytometry experiments, to be only to exosome enriched or total serum sample fractions (Figure 2D).

A representative example of our strategy of gating and confirmation of exosome identity. (A) We first identified singlets using FSC H versus FSC A analysis, then we identified our population of exosome-bound beads using SSC versus FSC analysis. The identity of this population was confirmed using analysis of the CD9 and CD81 staining; 2 different antibodies were used for these common markers for exosomes, and both antibodies were labeled with APC. (B) Western blot of the exosome marker CD63 using decreasing concentrations of expected exosomes. In parallel with the cytometry experiments, exosomes were isolated from serum, and CD63, the target of the antibody-coupled magnetic beads, was used to confirm the presence of exosomes in the samples, the band of interest is approximately 45 KDa. (C) Western blot of the exosome marker CD63 in total serum, exosome-free, and exosome-enriched samples. (D) Western blot of PD-L1 in total serum, exosome-free, and exosome-enriched samples.
We determined that 2 different wash steps, the first using centrifugation and the second using magnetic separation after the incubation with the antibodies, resulting in the highest number of antibody-positive events and the least background signal with the isotypes (Table 1).
Total Exosome Yield in Different Conditions.

We attempted to use 2 different magnets in the wash steps, the MojoSort and the BD IMag, and similar results for both were observed; following the protocol of the manufacturer, with 5 washes in total, more than 90% of the samples were lost. Then we attempted a no-wash protocol, where we obtained a great improvement in the number of events; however, this was accompanied by a very large increase in nonspecific background signal (ie, signals when only isotope controls, not the target antibody, were used, background signal which was in fact almost equal to the signal we saw when we used the target antibodies), which led us to try different numbers and types of wash steps. We used the final protocol (one centrifugation wash followed by IMag wash) for 20 samples and demonstrated improved yield over the other protocols (Table 1).
Next, after resolving the issue of the samples lost in the washing steps, the optimal starting concentration of exosomes was confirmed to be 200 µg per 200,000 beads. Concentrations ranging from 10 to 200 µg resulted in an antibody-positive yield in the bead:exosome region of approximately 20% to 80%. The antibodies used were for the classic exosome markers CD9 and CD81. In order to verify that the beads were binding intact whole exosomes, the exosome fraction was treated with ionic and nonionic detergents; SDS 1% was found to reduce the exosome signal to almost zero (Figure 3).
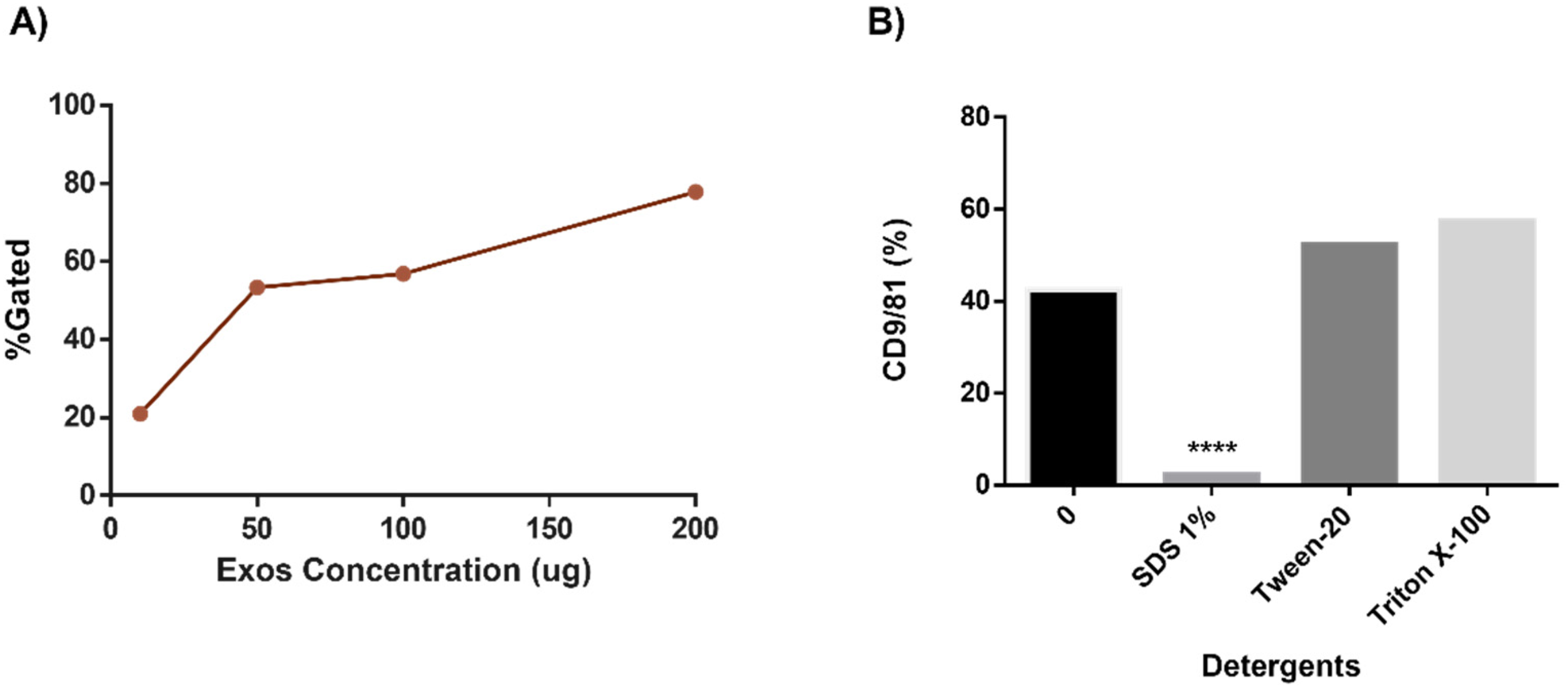
Effects of initial exosome concentration on flow cytometry final yield. (A) Different initial concentrations of exosomes (mg of protein) were tested in order to determine the conditions which lead to the highest percentages of markers within the exosome-bead gate which we had established (Figure 1); 200 mg gave the strongest signal. (B) Use of detergents to verify the presence of intact exosomes on the beads. As another control for our system, we tested ionic and nonionic detergents to determine which detergent was most effective at destroying the vesicles and to test whether the magnetic beads could capture fragments of exosomes and be detected by the cytometer.
This protocol was successfully used to identify immunoligands on exosomes. A representative experiment is shown in Figure 4. Here, we were able to identify the exosomes ligands that had been previously thought to exist only on the cell surface or in soluble forms, such as CD155 and PD-L1. The exosomes were positive for these ligands and the exosome markers CD9/81.

Example of the use of bead-based exosome isolation to identify relevant targets on exosomes. Measurement of immunoligands in exosome-positive beads. (A) Representative histograms of healthy donors and cancer patients, comparing their amount of PD-L1 with the isotype (PE), and a coexpression dot plot with CD9/81. (B) Representative histograms of healthy donors and cancer patients, comparing their amount of CD155 with the isotype (PE-Cy7), and a coexpression dot plot with CD9/81.
Immunodetection of Exosomes by Western Blot
As another control to confirm that we were working with exosomes isolated from serum, we detected through an immunodetection by SDS-PAGE Western Blot, in denaturing conditions. Bands with strong signals were visualized around 75 and 50 kDa in all the lanes, confirming the presence and correct isolation of the exosomes (Figure 2).
Cytometry of the Exosomes Coupled With the Magnetic Beads
This protocol allowed us to successfully identify over 80% of the beads in our defined region as being bound to exosomes, due to their identification through the classic markers CD9 and CD81. Additionally, we were able to identify cell surface ligands that had been previously thought to exist only on the cell surface or in soluble form, such as CD155 and PD-L1 (Figure 2 and Figure 4).
Discussion
In recent decades, the study of exosomes, and other EVs, has become increasingly popular and important in bioscience. Due to their presence in all types of cell and body fluids, the pivotal role that these structures play in intercell communication, and in the cell–cell delivery of a wide range of molecules, exosomes have attracted the focus of the scientific community. Exosomes have been proposed as potential drug carriers, important disease biomarkers, and therapeutic targets. Nonetheless, it has not yet been possible to reach a consensus on their correct handling, isolation, and detection, especially when using flow cytometry.
Although ultracentrifugation is the gold standard and the most common method used to isolate exosomes and other EVs from body fluids, it is very time and sample consuming. By definition, ultracentrifugation necessitates the need for an ultracentrifuge, and the high speeds, as well as the extended time commonly utilized in these protocols, could promote less than optimal purification via coupling between vesicles or with other cellular debris. This is why several companies have recently developed alternative exosome isolation kits that do not utilize ultracentrifugation. These kits have proven to be very suitable laboratory work, due to the fact that they are less time-consuming and require relatively small sample amounts. In fact, it has been reported that ultracentrifugation has a lower efficiency in the number of exosomes isolated from body fluids in comparison to some commercial kits, such as the kit from Invitrogen that we used in this study; additionally, these commercial kits often yield a very homogenous number and size of vesicles isolated and have been considered to be one of the better options to work with these EVs.16,17
As mentioned above, the commercial kit that we used was the Total Exosome Isolation Reagent from Serum, followed by the Exosome – Human CD63 Isolation/Detection Reagent (from Cell Culture Media). No commercial kit currently exists to couple exosomes from human serum to beads. This is primarily due to the fact that the serum samples have a large amount of various extraneous proteins, and that this commonly interferes with the coupling of the beads and exosomes. Due to this, we attempted to modify both manufacturer's protocols. Focusing first on the Total Exosome Isolation reagent, we used different times and forces of centrifugation in an attempt to clean our sample for use with the antiCD63 beads. We did not note any substantial difference resulting from our modifications and the suggested protocol, so we decided to continue with the standard suggested protocol. It is important to note that we verified the attempted modifications and the efficacy of the kit to the above protocol using Western blots for the CD63, a classic marker for exosomes. We found it necessary to continue the rest of the modifications in order to successfully use the beads optimized only for cell culture use with our serum samples.
Flow cytometry is currently one of the most used approaches in biomedical sciences; this is due to the fact that this technique provides multiparametric and quantitative information of roughly cell-sized objects in solution. 18 This technology has also been attempted to be applied to EVs; nevertheless, it has been necessary to modify existing cytometry in several aspects to fit the physicochemical characteristics of the exosomes. When working with exosomes, one can expect relatively low zeta potentials and refractive indexes compared with cells; this represents a major challenge. Additionally, in order to use flow cytometry to analyze exosomes, the instrument, and the protocol employed, must have the ability to resolve particles the size of exosomes (100 times smaller than the average mammalian cell), to reliably measure concentrations of such particles in a sample, and to detect and quantify their cargo. Together, these goals represent a great challenge. Commercial cytometers have a detection limit of around 500–200 nm, which makes it very difficult to correctly detect exosomes, which usually are around 50–150 nm. This small size, their origin, and their common co-precipitation with particles that can be autofluorescent, or nonspecifically bind antibodies, work together to potentially increase the background signal. Additionally, due to the abovementioned parameters, it is expected that the antibody-coupled signals in exosomes will be relatively dim as well. Among the adjustments one can make when attempting to optimize a protocol for exosome use in flow cytometry would be trying to decrease contamination with other particles in one's sample. This could be performed by filtration of all the fluids, such as the buffers and sheath fluid, used in the experiment. In order to improve the discrimination of singlets, a 2-fold serial dilution of the samples is recommended. Additionally, the use of a detergent treatment for the discrimination of nonvesicular (or fragments of exosomal membrane) events, as we did, is also recommended. 19
The majority of these examples of protocol adjustments are suitable mostly for a direct flow cytometry approach. Here, we have described the use of a bead-based approach. We used magnetic beads with a size suitable for resolution in commercial cytometers. These beads were covered with anti-CD63 capture antibodies. With this approach we were able to avoid the majority of the problems reported in the direct approach, nevertheless, one continuing issue remains: that is that even though we were detecting single events of beads + exosomes, due to the size of these, a single bead could have coupled to many heterogeneous exosomes. Despite this, the modification of the technique allowed us to detect and measure molecules bound to exosomes avoiding the vast majority of the complications experienced in direct flow cytometry.14,18–21
To a certain extent, our protocol might be said to only provide a relative, not absolute, quantification of the target proteins found on the exosomes. This is due to the fact that multiple exosomes can bind to each bead, however, we consider that the bead:exosome binding ratio would be relatively constant between experiments that used the same concentration of starting exosomes and the same concentration of beads. Thus, if a bead binds 10 or 100 exosomes, it will more or less constantly bind that number, and when using the same concentration of antibody in subsequent experiments, we can extrapolate antibody binding and fluorescence signal to the total exosomes and the differences between different samples. So, for example, if a PD-L1 negative sample has beads bound to 100 exosomes each, and a PDL1 positive sample has beads bound to 100 exosomes each, then we expect the PD-L1 positive samples to have a quantifiably higher signal than the PDL1 samples. This we did in fact see, so while the measurement is only relative here, we find it scales well with the conditions of the experiments, and in fact correlated all with our Western Blot analysis of multiple samples for PD-L1 (data not shown, part of another study): in fact this current protocol was developed in order to validate the results seen in densitometry analysis of Western Blot experiments, which are of course only semiquantitative.
With this modification to the flow cytometry of exosomes coupled to magnetic beads, we were able to successfully detect a population of exosomes positive for APC-labeled CD9/81. Not all CD63 beads are expected to be CD9/81 positives, for this reason, we do not have 100% agreement between results, having percentages of positivity from 60% to 91% between samples.
As another control for our system, we tested to see whether the magnetic beads could capture fragments of exosomes and be detected by the cytometer; the results seen in Figure 3B show that SDS detergent treatment of the exosome-enriched fraction essentially eliminated the yield of beads positive for exosome markers, thus reinforcing the idea that our protocol and cytometry gating strategy was giving us a readout of complete exosomes bound to beads, and not fractional parts of destroyed exosomes bound to beads.
Finally, as an example of the utility of this methodology, we were able to detect and measure these exosomes coupled to magnetic beads of 2 important immunoligands. PD-L1 is one of the major immune checkpoints due to its regulatory activity triggered by the coupling with its receptor PD-1. PD-L1 has been reported to have immunosuppressive activity, promoting tumor growth and inhibition of lymphocyte maturation and function, and due to this PD-L1 has become a promising target as a tumor biomarker and in the context of blockade therapies. Interestingly, the liberation and presence of PD-L1 on exosomes have been previously reported in cancers such as melanoma, lung, neck, and head cancer.10,12,22,23 In fact, even dimeric forms of PD-L1 have been reported bound to exosomes. 24 Although soluble, membrane, and bound to exosome forms of PD-L1 have never been compared, it is theorized that the exoPD-L1 could have stronger immunosuppressive activities compared to the other forms. 11 In light of these reports, our ability to use this protocol to identify and quantify PD-L1 on exosomes from the serum of cervical cancer patients represented a major technical advance for our group, and these results also validated data that we observed with western blots on the same exosome enriched samples. Similarly, we were able to identify CD155, which previously had been thought to exist only on the cell surface or in soluble forms.
Conclusion
This technique enables the quantitative evaluation of exosome proteins that had previously been difficult to reliably quantify. Additionally, our adapted protocol has allowed the identification of multiple proteins in what we assume is their native conformation, or very similar to it, on these exosomes. It is important to note that identification of proteins that are rarely expressed in exosomes is complicated in this technique as the serum is an inherently dirty and complicated source of exosomes, and great care must be taken in the washing and gating of the exosome:bead populations so as to not introduce noise, that would overshadow the experimental signal, to the system.
Footnotes
Abbreviations
Authors’ Note
Samples were obtained from women in different hospitals: Instituto de Cancerología, Hospital General de Occidente, and the Hospital Civil Nuevo de Guadalajara Juan I. Menchaca, where the protocol was approved by the Ethics Committees (CUCS: C.I.106/2016 IJC: PRO-20/17), and with informed consent signed by all participants, according to the World Medical Association Declaration of Helsinki, renewed in 2013 in Fortaleza, Brazil.
Acknowledgments
The authors thank Santa Cruz Biotech for generously providing us with several antibodies. AGAG acknowledges CONACYT for his PhD scholarship (886030).
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by a grant from the Consejo Nacional de Ciencia y Tecnología (CONACYT) through the Convocatoria “Fondo Sectorial de Investigación en Salud y Seguridad Social SS/IMSS/ISSSTE-CONACYT” (registration # 290195) to STA and by a grant from Universidad de Guadalajara through the Programa de Fortalecimiento de Institutos, Centros y Laboratorios de Investigación 2021 (P3E 260428, Clave CIP: lab-431 UdeG).