
Abstract
To explore the signature function of the tumor mutational burden (TMB) and potential biomarkers in prostate cancer (PCa), transcriptome profiles, somatic mutation data, and clinicopathologic feature information were downloaded from The Cancer Genome Atlas (TCGA) database. R software package was used to generate a waterfall plot to summarize the specific mutation information and calculate the TMB value of PCa. Least absolute shrinkage and selection operator (LASSO) Cox regression analysis was used to select the hub genes related to the TMB from the ImmPort network to build a risk score (RS) model to evaluate prognostic values and plot Kaplan–Meier (K-M) curves to predict PCa patients survival. The results showed that PCa patients with a high TMB exhibited higher infiltration of CD8+ T cells and CD4+ T cells and better overall survival (OS) than those with a low TMB. The anti-Mullerian hormone (AMH), baculoviral IAP repeat-containing 5 (BIRC5), and opoid receptor kappa 1 (OPRK1) genes were three hub genes and their copy number variation (CNV) was relatively likely to affect the infiltration of immune cells. Moreover, PCa patients with low AMH or BIRC5 expression had a longer survival time and lower cancer recurrence, while elevated AMH or BIRC5 expression favored PCa progression. In contrast, PCa patients with low OPRK1 expression had poorer OS in the early stage of PCa and a higher recurrent rate than those with high expression. Taken together, these results suggest that the TMB may be a promising prognostic biomarker for PCa and that AMH, OPRK1, and BIRC5 are hub genes affecting the TMB; AMH, OPRK1, and BIRC5 could serve as potential immunotherapeutic targets for PCa treatment.
Abbreviations
AMH, anti-Mullerian hormone; AMHRII, AMH type II receptors; BIRC5, baculoviral IAP repeat-containing 5; BPH, benign prostatic hyperplasia; CNV, copy number variation; DEGs, differentially expressed genes; DFS, disease-free survival; GEPIA, Gene Expression Profiling Interactive Analysis; GS, Gleason score; ICGC, International Cancer Genome Consortium; K-M, Kaplan–Meier; LASSO, least absolute shrinkage and selection operator; OPRK1, opioid receptor kappa 1; OS, overall survival; PCa, prostate cancer; PSA, prostate-specific antigen; rAMH, recombinant AMH; ROC, receiver operating characteristic; RS, risk score; SNP, single-nucleotide polymorphism; TCGA, The Cancer Genome Atlas; TGF-β, transforming growth factor beta; TIICs, tumor-infiltrating immune cells; TIM, tumor immune microenvironment; TMB, tumor mutational burden; TIMER, tumor Immune Estimation Resource.
Introduction
Prostate cancer (PCa) is the second most common malignant tumor in males and the fifth leading cause of cancer-related death worldwide, 1 although in recent years, some research progresses have been made in its pathogenesis and treatment.2–6 The latest report on cancer statistics estimated 174 650 new cases of PCa and 31 620 PCa-related deaths in the United States in 2019. 7 Currently, prostate-specific antigen (PSA) is the only commonly used circulating biomarker for the early diagnosis of PCa in the clinic. 8 However, benign prostatic hyperplasia (BPH) and prostatitis can also increase the serum PSA level, which creates certain limits for PSA screening. 9 Furthermore, the most common treatment for patients under 75 years old is radical prostatectomy and neoadjuvant endocrine therapy (castration or androgen blockade), 10 but up to 50% of patients suffer from biochemical recurrence after prostate removal.11,12 Therefore, it is of great significance to explore novel biomarkers and new therapeutic strategies for PCa treatment.
The immune system plays an essential role in cancer recognition and control. On the one hand, the immune system can protect the host from virus-induced tumors by clearing or suppressing viral infections. On the other hand, the immune system can specifically recognize tumor-specific antigens and destroy tumor cells before these cells cause damage, a process known as tumor-specific surveillance. 13 Thus, discovering the molecular determinants of immunotherapeutic responsiveness has been the focus of many efforts. 14 Currently, the neoantigen load and tumor-infiltrating lymphocytes are considered acknowledged molecular determinants.15,16 In addition, combinations of different immunotherapies have been explored to increase the efficiency of tumor immunotherapy. 17 The tumor mutational burden (TMB) has been considered a good predictor of immunological response and tumor behavior. 18 In recent studies, highly mutated tumors were shown to produce many novel peptides and thus more neoantigens that lead to increased tumor immunogenicity, rendering tumors more susceptible immune cell targets and resulting in an improved clinical response to immunotherapy.19,20 For example, patients with high-TMB ovarian cancer, skin cutaneous melanoma, or bladder urothelial carcinoma show a prolonged survival time, supporting the conclusion that a high-TMB may be a harbinger of good clinical outcomes. 21 Therefore, the TMB plays a vital role in immunotherapy and clinical prognosis in cancers. However, the function of the TMB in PCa patients and the identification of hub genes that affect the TMB are remain elusive.
In recent years, bioinformatics has provided a vital and effective tool for discovery in tumor-related research. Abundant somatic mutation data for tumor samples have been identified using whole-exome sequencing techniques. 22 Here, to investigate the correlations of the TMB with clinical prognosis and antitumor immunity in PCa, we compared and analyzed the gene sets of high-TMB and low-TMB groups of PCa patients based on The Cancer Genome Atlas (TCGA).
Materials and Methods
Data Collection
In this study, we downloaded somatic mutation data, a gene expression matrix, and clinical information for PCa samples from the TCGA. For the somatic mutation cohort, we first used the R GenVisR package to generate a waterfall plot to summarize the mutational data of the top 10 genes in the samples. We further used the “maftools” in R package to analyze the general characteristics of the somatic mutation data and comutation of the top 25 mutated genes. We subsequently calculated the TMB value (the total No. of mutations divided by the size of the target coding area) of each specimen. Finally, the mRNA expression matrix was obtained from the TCGA for later analysis.
Identification of Differentially Expressed Genes of the Tumor Mutational Burden Subgroups
We first divided the samples into a high-TMB group and a low-TMB group based on the median value of the TMB. Then, the “limma” R package was utilized to analyze differentially expressed genes (DEGs) between the two TMB groups. Specifically, the adjusted P value and |log2fold change| (|log FC|) were used to screen significant DEGs. We defined an adjusted P value < 0.05 and a |logFC| > 1 as the cutoff criteria.
Prediction of Immune Infiltration in Prostate Cancer Samples
CIBERSORT is an analytical tool that can provide precise values for different types of tumor-infiltrating immune cells (TIICs), which is based on a “gene signature matrix” of 547 genes. 23 In this study, we used DEGs expression to predict the content of 22 subtypes of immune cells per sample via the reference matrix provided by CIBERSORT. CIBERSORT was also used to calculate a reference P value to judge the accuracy of the TIIC data for each sample. A P value ≥ 0.05 was considered to indicate relatively low accuracy for prediction and filtered out of the matrix. On the contrary, a P value < 0.05 was viewed as accurate for prediction and retained.
Relationships between the Tumor Mutational Burden and Immune Cells
For the 22 subtypes of TIICs, we used violin plots and the “limma” R package to evaluate the difference in each immune cell type between the high-TMB group and the low-TMB group and created a violin plot to visualize the distributions of TIIC subtypes in the two groups. A P value < 0.05 was considered statistically significant.
Prognostic Analysis of the Tumor Mutational Burden
The International Cancer Genome Consortium (ICGC) database (https://icgc.org/) is a free public database. Data for 188 specimens (132 live patients and 56 dead patients) were downloaded from the ICGC. After calculating the TMB value and matching it with clinical survival data, we combined the TCGA clinical data with the ICGC data. Kaplan–Meier (K-M) curves were constructed to analyze the potential effects of the TMB on survival rates. We considered a P value < 0.05 statistically significant. We also utilized the “limma” R package to analyze the correlations between the TMB value and clinicopathologic features (eg age, T stage, and N stage).
Overlap between Differentially Expressed Genes and Immune Genes
The ImmPort ecosystem is an open-access dataset that includes more than 300 studies and is freely available at the Shared Data portal (www.immport.org/immport-open), which allows research data to be repurposed to accelerate the development of new insights into discoveries. 24 A total of 2498 immune-related genes were downloaded from ImmPort, and we identified overlapping genes between the DEGs and immune genes.
Construction of a Prostate Cancer Risk Score Model
To investigate the prognostic role of overlapping gene regulators in PCa, we utilized least absolute shrinkage and selection operator (LASSO) Cox regression analysis to screen hub genes from the overlapping genes. The hub genes and their coefficients were determined. Samples were separated into a high-risk group and a low-risk group based on the median risk score (RS) value and overall survival (OS) was evaluated. The OS prediction accuracy of the RS model was verified using a receiver operating characteristic (ROC) curve.
Influence of the Copy Number Variation of Hub Genes on the Immune Infiltrate in Prostate Cancer
tumor Immune Estimation Resource (TIMER, https://cistrome.shinyapps.io/timer/) is a comprehensive resource for systematic analysis of immune infiltrates across diverse cancer types and mainly contains six types of immune cells (eg B cells, CD4+ T cells, CD8+ T cells, neutrophils, macrophages, and dendritic cells) analyzed according to the TIMER algorithm to estimate cell infiltration. 25 We included hub genes in TIMER to explore the relationship between somatic copy number variation (CNV) and the abundance of TIICs in PCa.
Hub Genes Related to Clinical Features
After screening by LASSO Cox regression analysis, some hub genes were identified. We further probed the correlations of the hub genes with clinicopathologic features (eg age, T stage, and N stage) to study the influence of hub gene expression on clinical progression. With respect to the ability of hub genes to predict the OS and disease-free survival (DFS), we employed Gene Expression Profiling Interactive Analysis (GEPIA), which is a web-based tool that delivers fast and customizable functionalities based on TCGA data. 26
Results
Mutation Signature of The Cancer Genome Atlas Datasets
According to waterfall and maftools analysis by R package, we characterized various basic features of PCa somatic mutation data from the TCGA database (https://portal.gdc.cancer.gov). As identified by a waterfall plot, the top 10 mutated genes were TTN, TP53, SPOP, KMT2D, SYNE1, MUC16, FOXA1, KMT2C, SPTA1, and ATM. The clinical information showed that among 182 PCa patients, 62 (34.1%) were more than 65 years old, 154 (84.6%) were White, 24 (13.2%) were Black or African-American, and 4 (2.2%) were Asian (Figure 1A). The summary plot showed that the main variant classification was missense mutation, the major variant type was single-nucleotide polymorphism (SNP), and the most common type of simple nucleotide variation (SNV) class was cytosine changed into thymine (Figure 1B).

PCa mutation cohort in the TCGA. (A) Waterfall plot of the top 10 mutated genes in the TCGA PCa cohort. (B) Overview of mutations in the TCGA PCa cohort. (C) Volcano plot of DEGs between the high- and low-TMB groups. Upregulated genes are shown in green, downregulated genes are shown in red, and non-DEGs are shown in black.
Identification of Differentially Expressed Genes in Prostate Cancer
To explore the DEGs between high-TMB and low-TMB groups, the “limma” R package was applied for TCGA cohort analysis. The results demonstrated that a total of 185 DEGs were screened from the high-TMB and low-TMB groups, including 90 upregulated genes and 95 downregulated genes (Figure 1C).
Distribution of Tumor-infiltrating Immune Cells
To confirm the 22 subtypes of TIICs in PCa, we used CIBERSORT as an analytical tool to calculate the TIIC distribution in each PCa patient. The results revealed that the top three subtypes in the high-TMB group were resting memory CD4+ T cells, CD8+ T cells, and naive B cells, while in the low-TMB group, the top three subtypes were resting memory CD4+ T cells, naive B cells, and CD8+ T cells, respectively (Figure 2). Statistical analysis showed that compared to the low-TMB group, the high-TMB group had higher infiltration of CD8+ T cells (P < 0.05) and activated memory CD4+ T cells (Figure 2, P < 0.05), which may have vital roles in PCa.
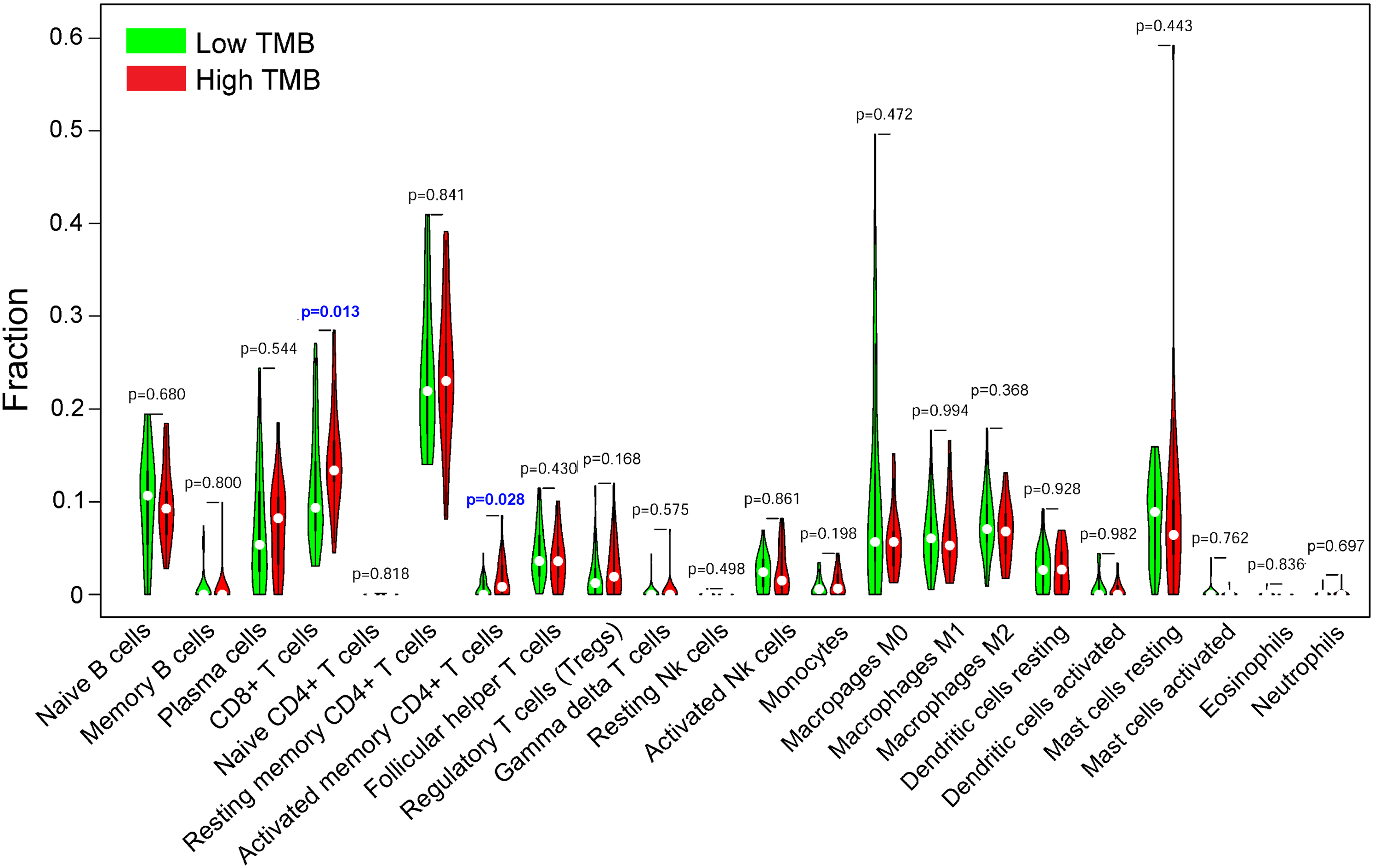
Differential analysis of 22 types of TIICs between the high- and low-TMB groups. CD8+ T cells and activated memory CD4+ T cells showed higher infiltration in the high-TMB group than in the low-TMB group (P < 0.05). There were no significant differences in the other 20 types of TIICs between the 2 TMB groups.
Survival Analysis and Clinicopathologic Features of the Tumor Mutational Burden
After processing with the survival analysis in R package, we built a K-M curve to determine the survival rate. The results showed significant differences between PCa patients with a low-TMB value and high-TMB value, and the patients with a low-TMB had poor OS (Figure 3A, P < 0.01). Moreover, compared with PCa patients younger than 65 years old, those older than 65 years old had a significantly higher TMB (Figure 3B, P < 0.01). In addition, we found that with increasing T and N stages, patients also had increasing TMB values (Figure 3C and D, P < 0.01). According to the K-M plot, a higher TMB favored longer survival, and we speculated that the high infiltration of CD8+ and CD4+ T cells might produce an antitumor effect. These data also suggested that a high TMB is closely related to an old age, a high T stage, and a high N stage, which coincides with the immune viewpoint that the more gene mutations there are, the more likely malignancy is.
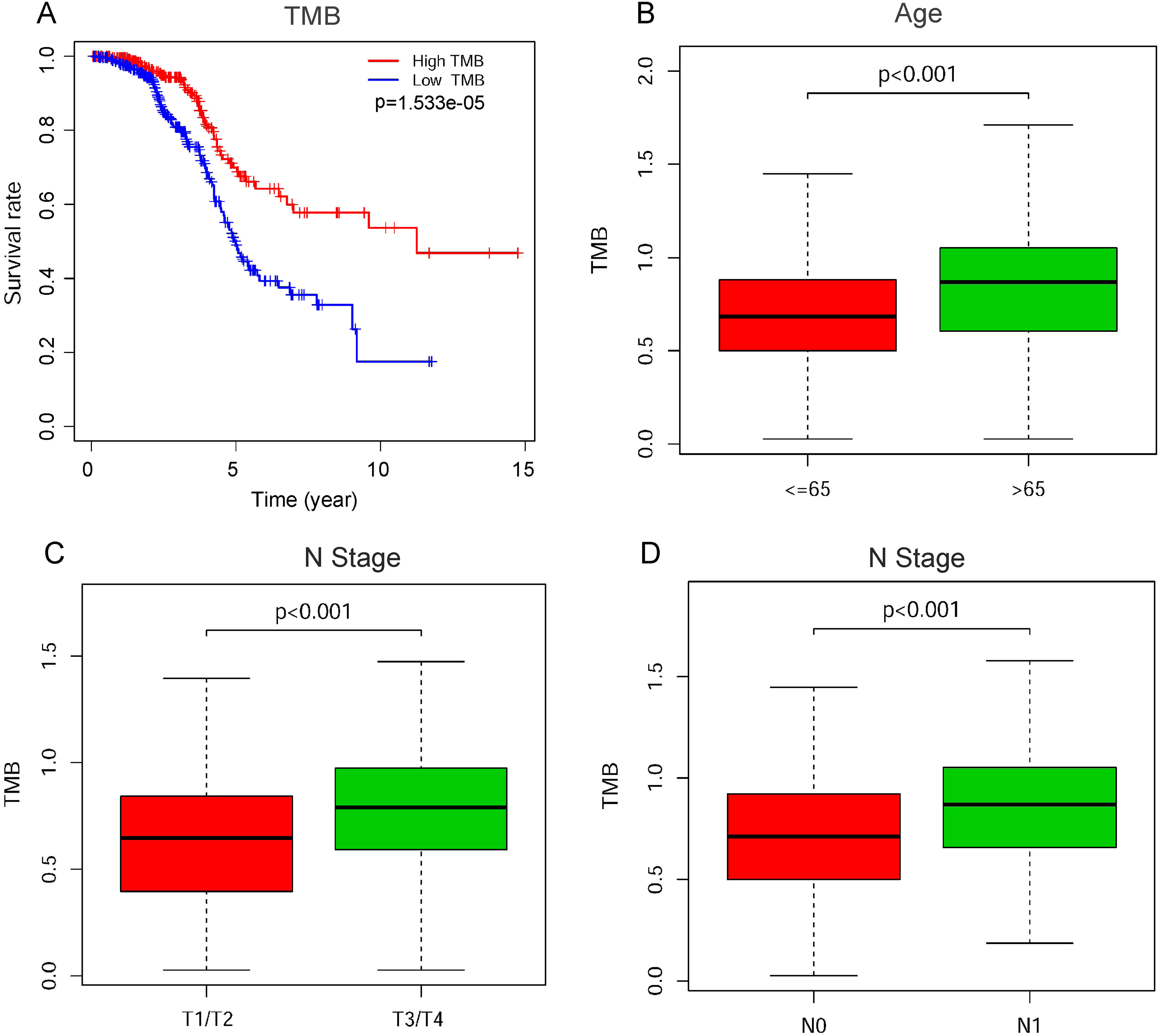
TMB-related clinical feature analysis. (A) Overall survival (OS) of the high- and low-TMB groups. The high-TMB group had longer survival than the low-TMB group (P < 0.001). (B) R software analysis of patients stratified by age. PCa patients older than 65 years had a significantly higher TMB than those younger than 65 years (P < 0.001). (C) R software analysis of patients stratified by T stage. The higher the T stage was, the higher the TMB value. (D) R software analysis of patients stratified by N stage. The TMB value of N1 patients was higher than that of N0 patients (P < 0.001).
Prostate Cancer Risk Score Model
After identifying the overlapping genes between DEGs and immune genes, we obtained 21 overlapping genes in total. We next used LASSO Cox regression analysis to screen the 21 overlapping genes and estimate the adjustment parameter λ by the cross-validation method. The results showed that when log (λ) = 5.5, the error rate was minimized (Figure 4A and B). After screening, three hub genes, AMH, OPRK1, and BIRC5, were retained to construct the RS model, in which the coefficients of AMH, OPRK1, and BIRC5 were 0.1857, −0.0442, and 0.0468, respectively. Then, we separated PCa patients into a high-risk group and a low-risk group based on the RS of each sample to investigate the prognostic role of the RS model. The results revealed that the high-risk PCa patients were predicted to have shorter OS times than the low-risk patients (Figure 4C, P < 0.05). Finally, a receiver ROC curve analysis was used to measure the accuracy of the RS model by determining the area under the ROC curve (AUC). The RS model precisely predicted the prognosis of the PCa patients (Figure 4D, AUC = 0.801).

Construction of the RS model for evaluating the prognostic value of three hub genes. (A) RS modeling by the LASSO Cox regression algorithm to screen 21 overlapping genes. (B) Distribution of the LASSO coefficients of the 21 overlapping genes. (D) Kaplan–Meier (K-M) plots of overall survival (OS) were generated with the RS model. (D) ROC curve evaluating the predictive efficiency of the RS model.
Influence of the Copy Number Variation of Hub Genes on Infiltrating Immune Cells in Prostate Cancer
The CNV of the three hub genes was investigated using the TIMER database. We found that deep deletion of AMH resulted in a low level of infiltrating CD8+ T cells in PCa compared with the normal copy number (Figure 5A, P < 0.05). Moreover, both the deep deletion and high amplification of AMH were associated with a low level of infiltrating CD4+ T cells in PCa compared with the normal copy number (Figure 5A, P < 0.05). For BIRC5, CD8+ T cells were found at a low infiltration level in PCa with a high amplification copy number compared with normal PCa (Figure 5B, P < 0.05), and CD4+ T cells were found to have a low infiltration level in PCa with a high amplification copy number compared with normal PCa (Figure 5B, P < 0.05), which indicated that the CNV of AMH, and BIRC5 could influence the infiltration level of CD8+ T cells and CD4+ T cells in PCa patients. However, the CNV of OPRK1 did not influence CD8+ or CD4+ T cell infiltration levels in PCa (Figure 5C, P > 0.05). The above analyses suggest that the CNV of AMH, BIRC5, and OPRK1 were relatively likely to affect the infiltration of immune cells in PCa, indicating that AMH, BIRC5, and OPRK1 could be potential immunotherapeutic targets for PCa treatment.

Effects of the CNV of AMH, BIRC5, and OPRK1 genes on immune cell infiltration. (A) The effect of the CNV of the AMH gene on six types of infiltrating immune cells in PCa. The CNV of the AMH gene relatively stronger affected the infiltration of CD8+ T cells, CD4+ T cells, macrophages, and neutrophils. (B) The effect of the CNV of the BIRC5 gene on six types of infiltrating immune cells in PCa. All six immune cell types were influenced by the CNV of the BIRC5 gene. (C) The effect of the CNV of the OPRK1 gene on six types of infiltrating immune cells in PCa. The CNV of the OPRK1 gene had a strong impact on macrophage infiltration.
Relationships between Hub Genes and Clinicopathologic Features
To clarify the clinical significance of the three hub genes in PCa prognosis, the OS and DFS were plotted as the survival curves by the GEPIA (http://gepia.cancer-pku.cn/) webtool. The OS results suggested that patients with low AMH expression had longer survival times than those with high AMH expression (Figure 6A, P < 0.05), and the DFS analysis indicated that elevated AMH expression favored disease progression in PCa (Figure 6B, P < 0.01). Similarly, PCa patients with low BIRC5 expression had longer OS (Figure 6C, P < 0.05) and DFS than those with high BIRC5 expression (Figure 6D, P < 0.01). Conversely, the survival analysis indicated that PCa patients with low OPRK1 expression had poorer OS (Figure 6E, P < 0.05) and longer DFS than those with high OPRK1 expression (Figure 6F, P < 0.05). These results indicate that AMH, OPRK1, and BIRC5 can serve as potential prognostic biomarkers in PCa.

K-M plots of the OS and DFS of PCa patients stratified by AMH, BIRC5, or OPRK1. (A–B) OS and DFS of AMH analyses based on AMH showed that high expression of AMH could shorten survival time and promote disease progression. (C–D) OS and DFS analyses based on BIRC5 showed that elevated BIRC5 expression could decrease the survival rates and promote disease progression. (E–F) OS analysis based on OPRK1 showed that high expression of OPRK1 might result in an elevated death rate in the early stage of PCa. DFS analysis based on OPRK1 showed that elevated OPRK1 expression might decrease the survival rate and increase tumor progression.
With respect to clinicopathologic features, no significant difference in age was found for any of the three hub genes (Figure 7A-C, P > 0.05). However, elevated expression of these three hub genes was associated with a high T stage (Figure 7D-F, P < 0.01). Similarly, AMH (Figure 7G, P < 0.01), OPRK1 (Figure 7H, P < 0.01), and BIRC5 (Figure 7I, P < 0.01) were more highly expressed in a high N stage than in a high T stage.

The relationship of the expression of the three hub genes and clinicopathologic features in PCa. (A–C) There were no significant differences in the expression of AMH, BIRC5, or OPRK1 between the two age groups (all P values > 0.05). (D–F) The expression levels of AMH, BIRC5, and OPRK1 were higher in T3/T4 than in T1/T2 (all P values < 0.001), which showed that the expression of the three hub genes might promote tumor progression. (G–I) The expression levels of AMH, BIRC5, and OPRK1 were higher in the N1 stage than in the N0 stage (all P values < 0.01), which revealed that the expression of the three hub genes might promote tumor metastasis.
Discussion
Despite great progress in diagnostic methods and therapeutic regimens, PCa remains a fatal condition for those who develop advanced disease. 27 TIICs are essential components of the tumor immune microenvironment (TIM), which can change the immune status of the tumor. Currently, studies have demonstrated that the TMB plays an important role in survival prognosis in several cancer types and may have a significant impact on the TIM.21,28 Targeted immunotherapeutic strategies have proven effects against various kinds of tumors.29–32 Therefore, exploration of whether the TMB can predict the prognosis of tumors and potential immunotherapeutic targets in PCa is urgently needed.
To our knowledge, this study is the first to correlate the TMB with clinical outcomes and immunotherapeutic targets in PCa by bioinformatics. In our study, mutation analysis of a TCGA dataset revealed that missense mutations, SNPs, and cytosine-to-thymine changes were the most common mutation types in PCa. We found that TP53 and SPOP were the most commonly mutated genes via a waterfall plot. Recent reports have suggested that TP53 mutation significantly correlates with a high Gleason score (GS) and PCa recurrence 33 and the accumulation of TP53 mutations increases T cell density in patients with PCa. 34 GS is one of the most important standards to determinate the tumor malignancy. In this study, we analyzed the relationship between TMB and GS, and found a significant positive correlation between them (Supplementary Figure). Additionally, SPOP mutation can promote prostate tumorigenesis via the PI3 K/mTOR and AR signaling pathways, 35 and SPOP mutation screening is a valuable personalized medicine tool for effective antitumor treatment. 36 Hence, both TP53 mutations and SPOP mutations may play important roles in PCa.
According to the somatic mutation profile, we identified and analyzed DEGs between the high-TMB group and the low-TMB group, and a total of 185 DEGs were identified. Based on these DEGs, 22 subtypes of TIICs were predicted per sample via the reference matrix provided by CIBERSORT. We found that CD8+ T cells and activated memory CD4+ T cells were more highly infiltrated in the high-TMB group than that in the low-TMB group. Traditionally, T lymphocytes, especially CD8+ cytotoxic T lymphocytes, are considered the main immunologic effector cells of antitumor immunity, as they can cause tumor cell death without affecting normal cells. tumor-associated T cells have been confirmed to improve the survival rate in multiple solid tumors independently.37,38 Moreover, T cell infiltration has a stronger independent effect on prognosis than current clinicopathological criteria, such as tumor size, depth of invasion, degree of differentiation, and lymph node status. 39 CD4+ T helper cells include Th1 cells, Th2 cells, Th17 cells, and regulatory T cells (Tregs) and are present in solid tumors at a frequency equal to or greater than that of CD8+ T cells. Meng et al. 40 suggested a positive association between activated memory CD4+ T cells and CD8+ T cells in PCa patients. Currently, increasing evidence emphasizes that CD4+ T cells play vital roles in antitumor immunity. 41 tumor infiltration by Th1 cells is associated with an improved survival rate in different types of cancer. 42 In addition, Th1 cells can generate cytokines, especially IL-2 and IFN-γ, that activate and promote the proliferation of CD8+ T cells and NK cells. 43 In the present study, based on the deconvolution algorithm CIBERSORT, we calculated the densities of 22 TIICs and compared them between the high-TMB group and the low-TMB group. We observed that CD8+ T cells and activated CD4+ T cells were at higher infiltrated levels in the high-TMB group. Additionally, according to our OS analysis based on the TMB, we found that a high-TMB had benefits for OS in PCa patients. Hence, we speculated that a high-TMB means more novel tumor antigens are generated, which makes the tumor vulnerable to attack and infiltration by immune cells 19 to lead to that PCa patients with an elevated TMB exhibit prolonged survival. It should be noted that tumor patients with high TMB are generally expected to have worse survival, whereas it is different from our findings in this study. For our result, one possible explanation is that higher TMB value signifies a higher heterogeneity of the tumor, which activates the body immune system to attack the tumor cells to benefit the patients. Some studies support our explanation, for example, Bi et al. 44 reported that a high TMB is associated with better clinical outcomes of ovarian cancer, and immune microenvironment analysis indicated the correlations between TMB and infiltrating immune cells. Zhang et al. 45 found that in bladder urothelial, carcinoma CD8+ T cell and memory-activated CD4+ T cell subsets not only revealed higher infiltrating abundance in high-TMB group but correlated with prolonged OS and lower risk of tumor recurrence, respectively. Together, our results coincided with the above immunological view and suggested that the TMB might be a potential prognostic factor in PCa that has potential value in targeted immunotherapy and may serve as a promising prognostic biomarker in PCa.
Anti-Mullerian hormone (AMH), a member of the transforming growth factor beta (TGF-β) family, is a potential therapeutic agent for tumor treatment.46,47 Both in vivo and in vitro studies have revealed that AMH can induce apoptosis and inhibit cancer cell growth in ovarian, breast, and PCa.48,49 AMH mainly induces cell apoptosis through specific AMH type II receptors (AMHRII), which are overexpressed in many cancer cells. Hence, recombinant AMH (rAMH) is viewed as a new potential anticancer agent, especially in diseases such as PCa. 50 AMH can also induce NF-κB signaling in PCa cells, 48 while NF-κB activity induced by dominant-negative IκB-α ablates AMH-mediated molecular events and inhibits PCa cell growth, suggesting that the prostate is a candidate target for the action of AMH. 51 In this study, high expression of AMH was observed in PCa with a high T stage, and overexpression of AMH was detected in PCa with a high N stage, which suggested that AMH had a high correlation with the clinical progression of PCa. We also found that the elevated AMH expression observed in PCa samples provided a better prognostic value for PCa patients. Collectively, the cancer pathogenesis and molecular mechanisms of AMH in PCa need to be assessed with in-depth studies.
Opioid receptor kappa 1 (OPRK1) is a member of the opioid receptors, G protein-coupled receptors that bind opioid ligands, including enkephalins and endorphins. As a tumor suppressor, an OPRK1 agonist was shown to reduce the growth of lung cancer. Xenograft experiments using mice showed that loss of OPRK1 enhanced melanoma and lung cancer tumor growth by suppressing angiogenesis. 52 Chen et al. 53 found that downregulation of the expression of the tumor suppressor κ-opioid receptor predicts a poor prognosis in hepatocellular carcinoma patients. Furthermore, a study on PCa performed by Hironobu et al. revealed that OPRK1 is an androgen-repressed gene that may suppress the growth of PCa. 54 Our study showed that a high level of OPRK1 in PCa samples was correlated with high-grade malignancy. Moreover, elevated OPRK1 expression levels in PCa showed potential roles as a prognostic biomarker in this cancer. Further studies should be carried out to explore the mechanisms underlying the function of OPRK1 during cancer pathogenesis.
Baculoviral IAP repeat-containing 5 (BIRC5) plays a crucial role in the occurrence and progression of cancer. 55 BIRC5 functions are involved in the tumorigenesis of colorectal tumors. 56 Moreover, Hu et al. considered BIRC5 to be a promising prognostic molecular biomarker in PCa. 57 Our study showed that elevated expression of BIRC5 not only had prognostic value in PCa but also presented close associations with clinical stage and tumor progression. Hence, the molecular mechanisms underlying the role of BIRC5 in human cancers require further research.
Footnotes
Acknowledgments
The authors would like to thank The Cancer Genome Atlas (TCGA), the International Cancer Genome Consortium (ICGC), the Shared Data portal, the ImmPort, the Tumor Immune Estimation Resource (TIMER), etc., databases listed in the manuscript for providing the data.
Authors’ Note
L.W. and Y.Y. contributed to the conceptualization and methodology, data collection, and writing the original draft. C.X. and X.W. contributed to statistical analyses, construction of the nomogram, and provision of the study materials. D.W. contributed to critical evaluation of the results. Z.H. contributed to revision of the manuscript and agreed to be accountable for all aspects of the work. All authors read and approved the final manuscript.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by the grants from the Shanghai Sailing Program (20YF1443800), the Science Foundation of Tongji Hospital (SKW2007), the National Natural Science Foundation of China (81671318), New Frontier Technology Joint Research Project of Shanghai Municipal Hospital (SHDC 12019112), and Clinical Research Plan of SHDC (SHDC2020CR3074B).
Ethical Statement
Our study did not require an ethical board approval because it did not contain human or animal trials.
Supplementa1 Material
Supplementary material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.