
Abstract
Neuroblastoma (NB) is a rare type of cancer but frequently occurred in children. However, it is still unclear whether circular RNAs (circRNAs) play key roles in NB tumorigenesis or progression. In this study, we identified 39,022 circRNAs across the 39 neuroblastoma and 2 normal cell lines. With the gene and circRNA expression data, we classified the NB cell lines, identified and characterized the functional circRNAs in the 3 NB classes. Specifically, 29 circRNAs were found to be dysregulated in the NB classes. Notably, 7 circRNAs located within MYCN-amplified regions were upregulated in cell lines with the high activities of MYC targets and MYCN amplification, and were highly correlated with expression of their parental gene, NBAS. Subsequently, we constructed ceRNA networks for the functional circRNAs. Specifically, hsa_circ_0005379 was identified as a critical regulator in the ceRNA networks because of targeting 13 genes, which formed a complex competing endogenous RNA (ceRNA) network. Moreover, hsa_circ_0002343, which was connected with few genes, might regulate the PI3K/Akt/mTOR signaling via RAC1. Furthermore, 3 genes, including NOTCH2, SERPINH1, and LAMC1, involved in epithelial mesenchymal transition (EMT) were observed to connect with hsa_circ_0001361, suggesting that this circRNA was closely associated with EMT. Consequently, 7 genes, such as DAD1, PPIA, NOTCH2, PGK1, BUB1, EIF2S1, and TCF7L2, were found to be closely associated with both event-free survival (EFS) and overall survival (OS). In conclusion, the present study identified functional circRNAs and predicted their functionality in neuroblastoma cell lines, which not only improved the understanding of circRNAs in neuroblastoma, but also provided the evidences for the related researchers.
Keywords
Introduction
Neuroblastoma (NB) is the most common solid tumor in childhood, which can be diagnosed using a combination of laboratory tests, radiographic imaging and pathology. 1 Several genetic alterations have been discovered in neuroblastoma cells, 2 including MYCN amplification, 11q loss of heterogeneity (LOH) or deletion, 1p loss, and point mutations in ALK and ATRX, which indicated an unfavorable prognosis. The recent advances in the molecular mechanism reveal that the initiation of NB may be caused by the somatic mutations during the embryonic development period, the frequency of which is relatively lower in NB than adult cancers. 3
In the past decade, more and more evidences show that noncoding RNAs (ncRNAs) can mediate cellular processes including chromatin remodeling, transcription, post-transcriptional modifications and signal transduction, 4 which improved our understanding that ncRNAs can act as functional regulatory molecules. Specifically, miR-204 was identified as a tumor suppressor microRNA (miRNA) that inhibited a subnetwork of oncogenes strongly associated with MYCN-amplified NB and poor outcome. 5 Moreover, several miRNAs such as microRNA-497, microRNA-34a, and miR-125b-1-3p were also identified to be involved in NB tumorigenesis or progression. 6 -9 Unlike the small size of miRNAs, the long noncoding RNAs (lncRNAs) were also found to drive NB tumorigenesis. For example, 2 lncRNAs located within 6p22 loci, CASC15 and NBAT1, were reported to act as tumor suppressors in NB. 10,11 Specifically, NBAT-1 controls neuroblastoma progression by regulating cell proliferation and neuronal differentiation. 11 However, the role of circular RNAs (circRNAs) in neuroblastoma is still unclear. In the present study, we for the first time detected the circular RNAs using RNA sequencing data, and investigated their associations with cancer-related pathways in NB. With the competing endogenous RNA (ceRNA) hypothesis, we identified circRNAs that potentially acted as ceRNAs and the mediating downstream pathways in NB, which provided circRNA evidences for related researchers.
Materials and Methods
Identification and Quantification of Circular RNAs in Neuroblastoma by RNA-Seq Data
The RNA-seq data of 39 neuroblastoma and 2 normal cell lines were downloaded from Sequence Read Archive (SRA) database with accession number SRP092413. 12 The 2 normal cell lines including hTERT-immortalized retinal pigmented epithelial cell line, RPE-1 (n = 1) and fetal brain (n = 1), were used as the normal control. The fastq files were aligned to human UCSC hg19 reference genome by BWA 13 0.7.12, and the circRNAs were identified by CIRI 14 v2.0. The read counts supporting the junction sites of the circRNAs were used as the expression levels of the circRNAs.
Gene Expression Quantification
The gene expression levels were estimated by hisat-stringtie-ballgown pipeline as proposed by previous study. 15 The abundances of the genes were quantified as Fragments Per Kilobase of transcript per Million (FPKM), and transformed to log2 (FPKM + 1).
Estimation of Pathway Activity
The cancer-related hallmark pathways were collected from the MSigDB 16 database. Pathway activities were estimated based on the log2 transformed gene expression data by single-sample gene set enrichment analysis 17 (ssGSEA).
CircRNA Differential Expression Analysis
The circRNAs detected in less than 10 cell lines were considered as lowly expressed and filtered. The DESeq2 method 18 was employed to identify the differentially expressed circRNAs based on the count-based expression matrix.
Predicting miRNA Targets
The miRNA-mRNA interactions were downloaded from the public database, 19 miRTarBase, which curated experimentally validated miRNA binding sites of mRNAs. The miRNA binding sites for circRNAs were predicted by miRanda 20 with default options.
Survival Analysis
The survival analysis was implemented in R with package survival. The Sequencing Quality Control (SEQC) neuroblastoma gene expression data were downloaded from Gene Expression Omnibus (GEO) database with accession number GSE49711. 21 The samples were stratified into high and low expression groups. The log rank test was used to test the survival difference between the 2 groups.
Results
Identification of Circular RNAs in Neuroblastoma Cell Lines
We collected RNA sequencing data of 39 neuroblastoma (NB) and 2 normal cell lines from publicly available dataset with SRA accession number SRP092413, the cDNA libraries of which were constructed by Ribo-Zero technology, allowing us to identify the circular RNAs in these samples. Totally, we identified 39,022 circRNAs across the 41 samples (read count > 1), and found 2 circRNA hotspots at 2p and 14q (Figure 1A). Particularly, the circRNAs located within the hotspot at 2p were originated from the genes adjacent to MYCN, a well-recognized oncogene amplified in NB, suggesting that frequent detection of these circRNAs might be resulted from the MYCN amplification.
The normalization of circRNA counts by the chromosome length revealed circRNAs might be more frequently transcribed from the chromosomes 17, 19, 2, 12, and 14, and rarely transcribed from chromosomes Y, X, 4, 21, and 13 (Figure 1B). When annotated by gene models, the circRNAs were found to be principally originated from exonic regions (84.69%), followed by intronic and intergenic regions (Figure 1C). Furthermore, we also investigated whether the circRNA expression was affected by their host genes. Overall, correlation between the expression medians of circRNAs and their host genes was insignificant (Figure 1D), suggesting that circRNA expression was regulated by multiple processes, not only transcription.

The overview of the circRNAs in neuroblastoma cell lines. (A) The genome-wide visualization of circRNAs identified by RNA-seq. The bars present the number of circRNAs located within this region. (B) The number of circRNAs per million bases (MBases) on the chromosomes. The circRNA count was normalized by dividing their chromosome length. (C) The proportion of exonic, intronic, and intergenic circRNAs. (D) The expression levels of circRNAs and their parental genes.
Identification of Functional circRNAs Involved in Neuroblastoma
To identify the functional circRNAs, we first characterized the dysregulated pathways for the 39 neuroblastoma (NB) cell lines by single-sample gene set enrichment analysis (ssGSEA). We observed that the 39 NB cell lines could be clustered into 3 classes by hierarchical clustering analysis (Figure 2A), with 12, 9, 18 cell lines, respectively. Notably, the classes II and III were shown to have higher activities of E2F targets and MYC targets, suggesting that the MYCN amplification might be the major cause of dysregulation of the 3 pathways in these cell lines. However, MYCN amplification was also observed in most of the cell lines, but lower proportion in class I (11/18, 61%). Moreover, Hedgehog signaling was observed upregulated in class I. In addition, the metastasis-related pathways, such as TGF-beta signaling, angiogenesis, and epithelial mesenchymal transition (EMT), were upregulated in class III and a small proportion of cell lines of class I.
With the classification for NB cell lines, we conducted pairwise differential expression analysis of the circRNAs stably expressed in the NB cell lines. Totally, 29 circRNAs were found to be dysregulated between the 3 classes (Figure 2B, P-value < 0.001 and fold change >2). Notably, the circRNAs located within MYCN-amplified regions, such as hsa_circ_0003287, hsa_circ_0008083, hsa_circ_0052767, hsa_circ_0000978, hsa_circ_0117720, chr2:15467874|15567918, and hsa_circ_0008261, were upregulated in class II, which were consistent with the high activities of MYC targets in class II. These results suggested that these dysregulated circRNAs might be involved in the tumorigenesis or progression of neuroblastoma.
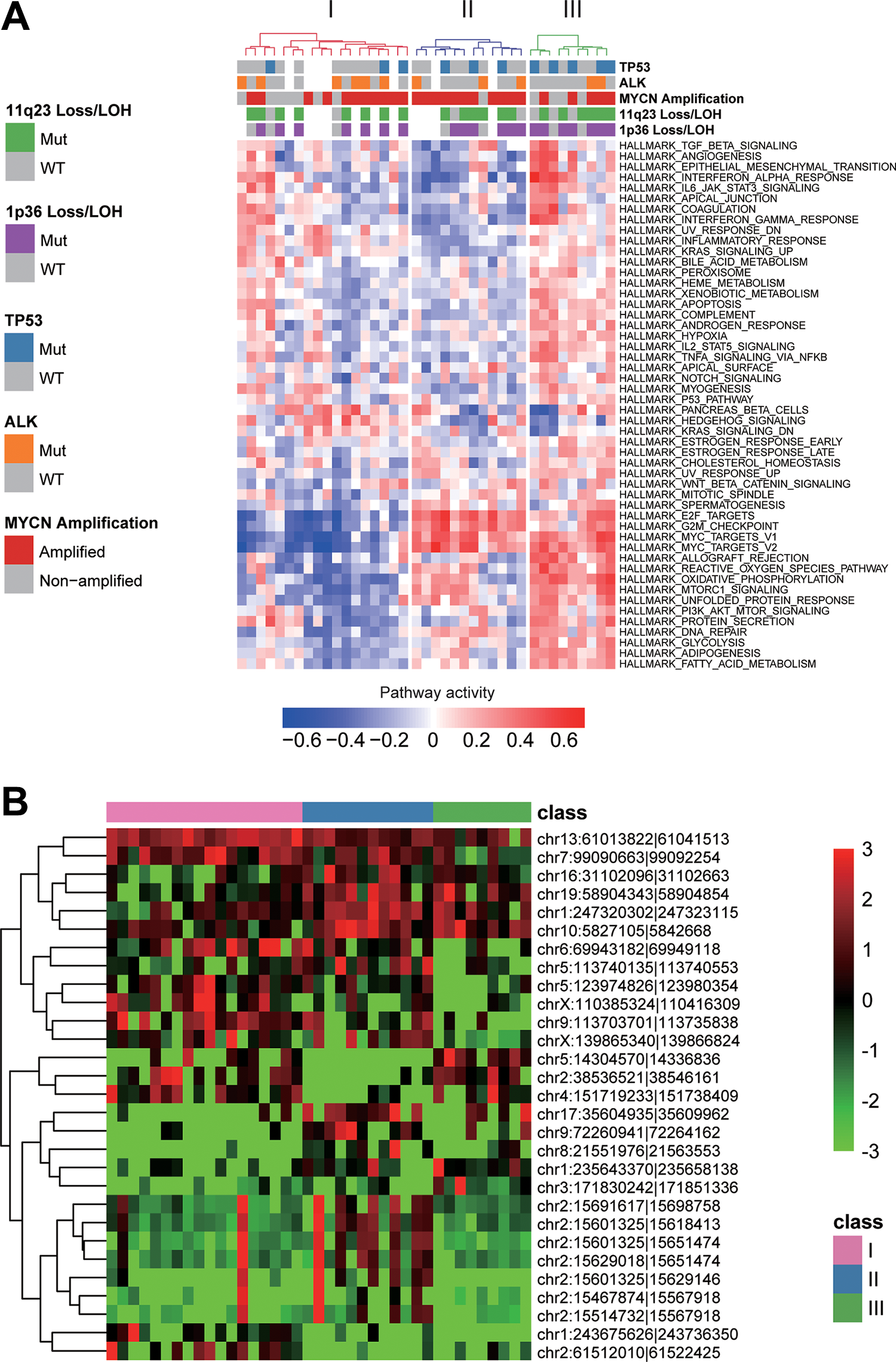
Neuroblastoma classes and class-specific pathways and circRNAs. (A) The 3 classes stratified by the pathway activities estimated by single-sample gene set enrichment analysis (ssGSEA). (B) The class-specific circRNAs by pairwise differential expression analysis.
Parental Genes of the circRNAs Involved in Neuroblastoma
As circRNAs have been found to promote or inhibit transcription of their parental genes, 22 -25 we first examined their associations with corresponding parental genes. As shown in Figure 3A, all of the functional circRNAs had positive correlation with their parental genes except hsa_circ_0008618. Compared with the insignificant correlation in all circRNAs, the functional circRNAs exhibited high correlation with their parental genes. Remarkably, 7 circRNAs transcribed from NBAS, a neuroblastoma amplified sequence gene adjacent to MYCN, were highly correlated with NBAS expression (Pearson correlation coefficient, PCC > 0.4), further suggesting dysregulation of these circRNAs might be caused by NBAS gene amplification. In addition to NBAS, the remaining circRNAs were not observed to produce circRNA isoforms.
Furthermore, the potential signaling pathways that circRNAs were involved in were predicted by the correlation analysis. Given a threshold of 0.5 for the PCCs between circRNAs and pathways, 10 circRNAs were successfully predicted to participate in over 1 pathway (Figure 3B). Particularly, hsa_circ_0002343, hsa_circ_0005379, hsa_circ_0001361, and hsa_circ_0084606 were found correlated with multiple pathways (n > 5). Moreover, hsa_circ_0003611, hsa_circ_0003441, hsa_circ_0000280 were predicted to participate in only 1 pathway, Hedgehog signaling. These results indicated that circRNAs might have multiple biological functions and multiple circRNAs might also be jointly involved in the same pathway. Further analysis revealed that none of the host genes had been found to participate in these pathways, giving us a hint that the circRNAs participated in these pathways not via their parental genes.

The correlation between circRNAs and parental genes. (A) The Pearson correlation coefficients (PCC) between the circRNAs and parental genes were plotted as the nodes. (B) The circRNAs and their mediating pathways predicted by correlation analysis.
The circRNA-Mediated Competing Endogenous RNA Network
As the noncoding RNAs like long noncoding RNAs and circRNAs could act as microRNA sponges to regulate expression of the downstream genes, we then searched for the miRNA binding sites of the circRNAs. It should be noted that the miRNA binding sites must cover the 3 bases at the 2 flanks of the junction site. Moreover, the target genes of circRNAs and the circRNAs should participate in the same pathway. Among the functional circRNAs, 4 were predicted to have the potential to compete with the 21 mRNAs for the miRNAs (Figure 4). Particularly, hsa_circ_0005379 was predicted to regulate 13 target genes, which formed a complex competing endogenous RNA (ceRNA) network. In addition, hsa_circ_0002343 and hsa_circ_0005598 were found to regulate fewer target genes. Specifically, hsa_circ_0002343 might regulate the PI3K/Akt/mTOR signaling via RAC1. The pathways like MYC targets and mTORC signaling were frequently predicted to be regulated by the ceRNA network. Furthermore, 3 genes involved in epithelial mesenchymal transition (EMT) were observed to connect with hsa_circ_0001361, suggesting that this circRNA was closely associated with EMT.
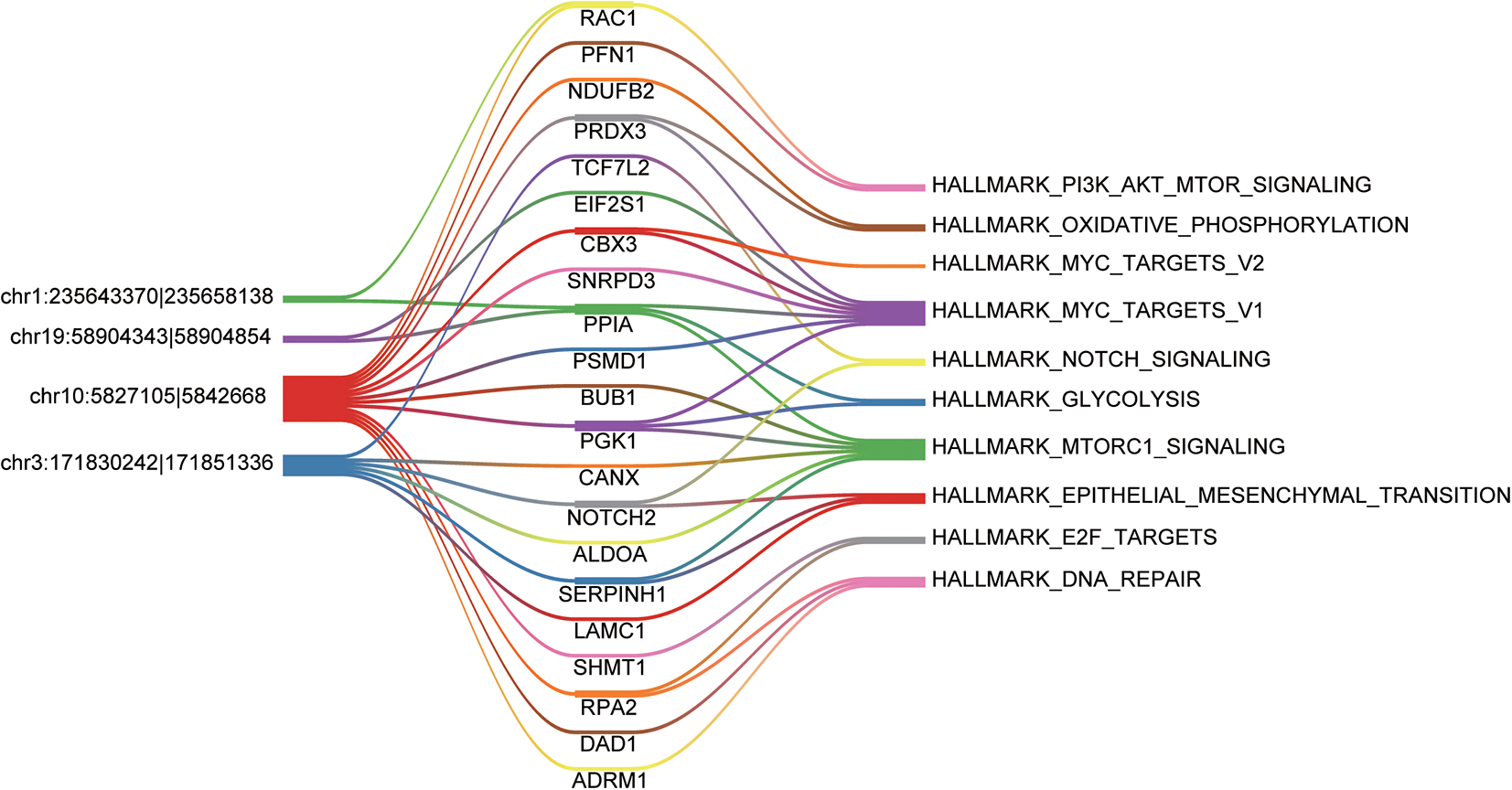
The competing endogenous RNA (ceRNA) network. The color bands connected the circRNA-mRNA and mRNA-pathway relationships.
The Prognostic Values of the circRNA Targets in Neuroblastoma
The target genes in the ceRNA network were subjected to the univariable Cox regression analysis based on the gene expression data from Sequencing Quality Control (SEQC) 26 (Sequencing Quality Control) cohort (n = 498). Remarkably, 12 of the 21 target genes were observed highly correlated with both event-free survival (EFS) and overall survival (OS) (Figure 5A and B, P-value < 0.05), of which, NOTCH2 and TCF7L2 were positively correlated with EFS and OS, suggesting that their expression indicated a favorable outcome.
Moreover, a stepwise method was used to select these genes in EFS- and OS-based multivariable Cox models. Specifically, 7 genes, such as DAD1, PPIA, NOTCH2, PGK1, BUB1, EIF2S1, and TCF7L2, were selected for construction of either the EFS- or OS-based Cox model (Supplementary Table S1 and Table 1). Particularly, DAD1, PGK1, BUB1, and TCF7L2 were included in both of the Cox models. The 498 samples were then stratified into high-risk (HR) and low-risk (LR) groups based on the risk scores estimated by the EFS- or OS-based Cox model. As shown in Figure 5C-D, the EFS-HR and OS-HR groups showed significantly higher risk of poor prognosis than the EFS-LR and OS-LR groups (log-rank test, P < 0.0001), respectively. These results indicated that the target genes mediated by the circRNAs might be indicators of NB prognosis.
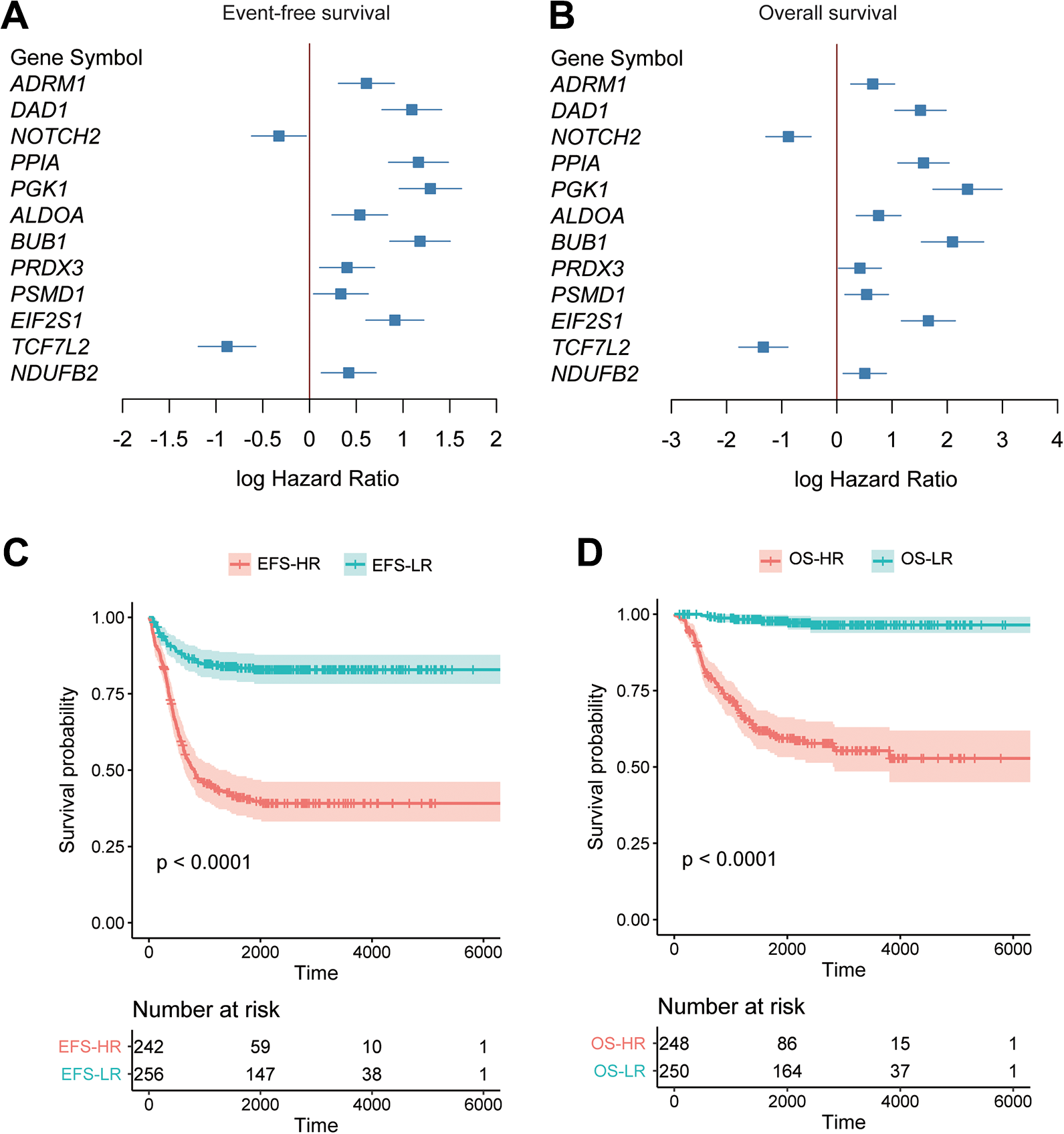
The prognostic values of the circRNA target genes. The circRNA target genes with significant correlation with both event-free survival (EFS) and overall survival (OS) were displayed in (A) and (B). The Cox models were constructed based on the circRNA target genes, and the samples were stratified into high-risk and low-risk groups. Kaplan-Meier (KM) curves for the EFS-HR vs. EFS-LR and OS-HR vs. OS-LR were plotted in (C) and (D).
The Coefficients and Statistical Significance of the circRNA Target Genes in Event-Free Survival and Overall Survival Based Multivariable Cox Models.

Discussion
Neuroblastoma (NB) is a rare type of cancer but frequently occurred in children. The circRNAs are implicated in cancers; however, it is still unclear whether circRNAs play key roles in NB tumorigenesis or progression.
In this study, we identified 39,022 circRNAs across the 39 neuroblastoma and 2 normal cell lines (read count > 1). Particularly, the circRNAs located within the hotspot at 2p were originated from the genes adjacent to MYCN, a well-recognized oncogene amplified in NB, suggesting that frequent detection of these circRNAs might be resulted from the MYCN amplification. With the gene and circRNA expression data, we attempted to classify the NB cell lines, identify and characterize the functional circRNAs. Specifically, 29 circRNAs were found to be dysregulated in NB. Notably, the circRNAs located within MYCN-amplified regions, such as hsa_circ_0003287, hsa_circ_0008083, hsa_circ_0052767, hsa_circ_0000978, hsa_circ_0117720, chr2:15467874|15567918, and hsa_circ_0008261, were upregulated in cell lines with the high activities of MYC targets and MYCN amplification. The ncRNAs transcribed from the copy number alteration (CNA) regions were frequently reported as cancer drivers. 27,28 Similarly, the 7 circRNAs within MYCN region might also have the potential to drive NB initiation.
As circRNAs have been found to promote or inhibit transcription of their parental genes, 22 -25 we found the functional circRNAs had positive correlation with their parental genes. Consistently, the 7 circRNAs located within MYCN-amplified regions were transcribed from NBAS, a neuroblastoma amplified sequence gene adjacent to MYCN, and were highly correlated with NBAS expression (Pearson correlation coefficient, PCC > 0.4). NBAS was also identified as an unfavorable indicator of NB due to its co-amplification with MYCN, 29 but its functionality in NB is not quite clear. Based on the correlation analysis of the circRNAs and pathway activities, we found that none of the parental genes was involved in the pathway highly correlated with circRNAs, giving us a hint that the circRNAs participated in these pathways not via their parental genes. Subsequently, we constructed ceRNA networks for the functional circRNAs. Specifically, hsa_circ_0005379 was identified as a critical regulator in the ceRNA networks because of targeting 13 genes, which formed a complex competing endogenous RNA (ceRNA) network. Moreover, hsa_circ_0002343, which was connected with few genes, might regulate the PI3K/Akt/mTOR signaling via RAC1. RAC1 and PI3K/Akt/mTOR signaling played key roles in neuroblastoma. 30,31 Furthermore, 3 genes involved in epithelial mesenchymal transition (EMT) were observed to connect with hsa_circ_0001361, suggesting that this circRNA was closely associated with EMT. Furthermore, EMT was mediated by Polo-like kinase 4 (PLK4) via PI3K/Akt signaling in neuroblastoma. 32 Similarly, we speculated that EMT might be mediated by hsa_circ_0001361 via NOTCH2, SERPINH1, and LAMC1.
As the lack of circRNA expression data with long-term follow-up, we indirectly investigated the prognostic values of the target genes instead of the functional circRNAs. Seven genes, such as DAD1, PPIA, NOTCH2, PGK1, BUB1, EIF2S1, and TCF7L2, were found to be closely associated with both EFS and OS. Functionally, genes such as NOTCH2, PGK1, and BUB1 have been reported to promote or suppress the neuroblastoma progression. 33 -35 Prognostically, PGK1 was identified as a predictor of neuroblastoma outcome. 36
In conclusion, the present study identified functional circRNAs and predicted their functionality in neuroblastoma cell lines, which not only improved the understanding of circRNAs in neuroblastoma, but also provided the evidences for the related researchers.
Supplemental Material
Supplemental Material, Supplementary_Table_S1 - Comprehensive Characterization of Circular RNAs in Neuroblastoma Cell Lines
Supplemental Material, Supplementary_Table_S1 for Comprehensive Characterization of Circular RNAs in Neuroblastoma Cell Lines by Li Zhang, Hangyu Zhou, Jing Li, Xinyu Wang, Xin Zhang, Tieliu Shi and Guoshuang Feng in Technology in Cancer Research & Treatment
Footnotes
Abbreviations
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by Beihang University & Capital Medical University Advanced Innovation Center for Big Data-Based Precision Medicine Plan (BHME-201801).
Supplemental Material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.