
Abstract
Objectives:
Bortezomib has been widely used to treat multiple myeloma and other hematological malignancies. However, not much is known about its effect on solid tumors. The aim of this study was to study the effect of Bortezomib on human esophageal cancer cell lines and investigate the potential target pathways.
Methods:
Two human esophageal cancer cell lines, TE-1 and KYSE-150, were used in this study. Cell viability, cell cycle distribution, and apoptosis after Bortezomib treatment was detected by Cell Counting Kit-8, flow cytometry, and Annexin V/propidium iodide staining, respectively. The genes targeted by Bortezomib were analyzed at the messenger RNA level by microarray chips and quantitative real-time polymerase chain reaction.
Results:
The proliferation of human esophageal cancer cell lines was inhibited by Bortezomib in a dose- and time-dependent manner. Bortezomib treatment led to G2/M arrest and apoptosis. Microarray chips revealed multiple signaling pathways targeted by Bortezomib, including proteasome, endoplasmic reticulum, Wnt-, and calcium-mediated pathway. The expression patterns of 4 representative genes UBD, CUL3, HDAC6, and GADD45A were verified by quantitative real-time polymerase chain reaction and showed consistency with the microarray assay.
Conclusion:
Bortezomib could suppress cell viability, cause G2/M arrest, and induce apoptosis in human esophageal cancer cells, with possible targets including UBD, CUL3, HDAC6, and GADD45A.
Introduction
Esophageal cancer ranks as the sixth leading cause of cancer-related deaths worldwide. 1 Esophageal squamous cell carcinoma (ESCC) is the predominant histological type of esophageal cancer in the East Asian population. Due to the lack of symptoms, esophageal malignancies have a particularly poor prognosis. More than half of the patients are diagnosed with distant metastases or unresectable disease, leading to a dismal <20% 5-year survival rate. Chemotherapy along with or followed by radiotherapy is the standard therapeutic approach for patients with late-stage ESCC. 2 Hence, developing novel and more effective chemotherapeutic agents has become the main focus in ESCC treatment.
Proteasomes play an important role in regulating the cellular activities such as cell cycle, cell proliferation, and apoptosis and often show resistance to chemo- and radiotherapies. 3 Since cancer cells are highly sensitive to altered proteasome activity when compared to normal cells, 4 proteasomes have been investigated as novel molecular targets in cancer treatment.
Bortezomib (Velcade, MG-341, PS-341), a potent 26S proteasome inhibitor, was the first of its kind to be approved by the Food and Drug Administration in 2003 for treating multiple myeloma (MM). Although the clinical antitumor activity of Bortezomib is well established in various hematological malignancies 5 -7 and several solid tumors, 8,9 the therapeutic effects of Bortezomib on ESCC have been far less impressive. Several studies indicated the therapeutic effects of the combination of Bortezomib with other antitumor therapy (eg, chemotherapy, immunotherapy, or radiotherapy). 10 -13 However, the effect of Bortezomib on esophageal cancer cells as a single agent and its underlying molecular mechanism is still not fully understood.
In this study, we tested the effect of Bortezomib on esophageal cancer cells as a single agent and thoroughly explore the underlying mechanisms responsible for Bortezomib-mediated cytotoxicity by microarray chips. The viability of the cancer cells was negatively affected by Bortezomib in a dose- and time-dependent manner. Bortezomib treatment led to both G2/M arrest and apoptosis in the cell lines. Microarray chip data revealed certain possible targets such as UBD, CUL3, HDAC6, and GADD45A, suggesting that Bortezomib is able to affect multiple signaling pathways including proteasome, endoplasmic reticulum (ER), Wnt-, and calcium-mediated pathway.
Materials and Methods
Cell Culture
Esophageal carcinoma cell lines TE-1 and KYSE-150 were purchased from the Biochemistry and Cell biology Institute of Shanghai, Chinese Academy of Sciences. Both cell lines were maintained at 37°C, 5% CO2 in Dulbecco’s Modified Eagle’s Medium (Hyclone; GE, China) supplemented with 2 mmol/L
Cell Proliferation Assay by Cell Counting Kit 8
Cell Counting Kit 8 (CCK-8; Dojindo Laboratories, Japan) was used to assess the cell viability according to the manufacturer’s instructions. Briefly, 5 × 103 cells were seeded per well with 100 µL medium in a 96-well plate and incubated overnight. The cells were then treated with different concentration of Bortezomib for the stipulated duration, followed by the addition of 10 μL of the CCK-8 solution. Two hours later, the absorbance values (OD450) were measured using a VersaMax Microplate Reader (BioTek, Winooski, Vermont). All experiments were setup in quintuplicate, and the data are presented as the mean (standard deviation [SD]).
Cell Cycle Progression Analysis
Cells were seeded at the density of 1.0 × 105 per well in a 6-well plate in triplicates and cultured overnight before being treated with Bortezomib. Cells were harvested 24 hours later, resuspended in 0.3 mL of 0.5% FBS in phosphate-buffered saline (PBS) and 0.7 mL 100% cold ethanol, and fixed overnight at −20°C. The fixed cells were resuspended in 800 μL PBS, washed once with 1 mL ice-cold PBS, and then incubated with 200 μL of 0.25 mg/mL RNase (Sigma, St Louis, Missouri) at 37°C for 30 minutes. Following incubation, the cells were stained with 300 μL of 0.1 mg/mL propidium iodide (PI; Sigma, St Louis, Missouri) for 15 minutes in the dark. Cell cycle distribution and DNA content were then measured using a flow cytometer (FACSVerse; BD, San Diego, California).
Apoptosis Assay
The apoptotic ratios of cells were determined with the FITC Annexin V Apoptosis Detection Kit I (BD Pharmingen, San Diego, California). A total of 1 × 105 cells per well in a 6-well plate were treated in triplicates and harvested 48 hours afterward. The cells were washed, resuspended in 100 μL of 1× binding buffer, and 5 μL each of FITC Annexin V and PI were added to the cells. After gently vortexing, the cells were incubated for 15 minutes at room temperature in the dark, and the reaction was stopped by adding 400 μL of 1× binding buffer per tube per manufacturer’s instructions. The cells were examined within 1 hour on an FACSVerse (BD, San Diego, California) flow cytometer, and the data were analyzed with Flow software (BD, San Diego, California).
Protein Extraction and Western Blot
Protein extraction and Western blot were performed according to published methods. 14 The following primary antibodies were used: cyclin B1, cleaved caspase-3 (Cell Signaling Technology, Beverly, Massachusetts), and β-actin (Beyotime Institute of Biotechnology, Haimen, China). All primary antibodies were used at a dilution of 1000-fold.
Microarray Analysis
The TE-1 cells were seeded in 10-cm dishes and treated with 150 nM Bortezomib or an equivalent volume of vehicle in triplicates. After 24 hours of incubation, the cells were harvested, and their total RNA was extracted for gene expression assays. Gene expression profiling was performed using Whole Human Genome Oligo Microarray v2 (4 × 44 K; Agilent Technologies, Santa Clara, California) which contained approximately 44 000 assay probes corresponding to all of the annotated human messenger RNA (mRNA) sequences (from RefSeq Build 36.3, Ensemble Release 52, Unigene Build 216 and GenBank, April 2009). Total RNA labeling and array hybridization were performed using standard protocols according to the manufacturers’ instructions. The Agilent Scanner G2505C (Agilent Technologies, Santa Clara, California) was used to scan the probe arrays, and Agilent Feature Extraction software (version 11.0.1.1) was used to analyze acquired array images. Quantile normalization and subsequent data processing were performed using the GeneSpring GX v11.5 software package (Agilent Technologies, Santa Clara, California). After quantile normalization of the raw data, genes that in least 3 of 6 samples had flags in “Detected” (“All Targets Value”) were chosen for further data analysis. Differentially expressed genes with statistical significance were identified through Volcano Plot filtering, and hierarchical clustering was performed using R. Gene Ontology analysis and pathway analysis were performed using the standard enrichment computation method.
RNA Isolation
Cells were seeded in 10-cm dishes at the density of 1.0 × 106 in triplicates and cultured overnight before Bortezomib treatment. After 24 hours of incubation, the cells were harvested and their total RNA extracted with RNAiso Plus (TAKARA, Otsu, Japan) according to the manufacturer’s protocol. The RNA was quantified using a NanoDrop ND-1000 (Thermo Scientific Inc, Waltham, Massachusetts), and its integrity was assessed by standard denaturing agarose gel electrophoresis.
Quantitative Real-Time Polymerase Chain Reaction Assay
Extracted RNA was reverse transcribed to complementary DNA (cDNA) using PrimeScript RT reagent Kit (Perfect Real Time; TAKARA, Otsu, Japan) according to the manufacturer’s protocol. The SYBR Green Real-Time Polymerase Chain Reaction (PCR) assay kit (TAKARA, Otsu, Japan) was used for amplification of cDNA. The mRNA levels of UBD, CUL3, HDAC6, and GADD45A, as well as that of the internal standard glyceraldehyde 3-phosphate dehydrogenase, were measured by real-time PCR in triplicates using a 7500 Real-Time PCR system (Applied Biosystems, Foster City, California). Primers specific to the abovementioned genes are listed in Table 1.
Primer Sequences for Real-Time PCR Analysis.

Abbreviations: GAPDH, glyceraldehyde 3-phosphate dehydrogenase; PCR, polymerase chain reaction.
Statistical Analysis
Mean values (SD) were used for data presentation. Student t test was used for 2-group comparison, and 1-way analysis of variance was used for more than a 2-group comparison by GraphPad Prism 5.0 Software. A P value <.05 was considered to imply a statistically significant difference.
Results
Bortezomib Inhibits the Proliferation in Esophageal Carcinoma Cells
To examine the effect of Bortezomib on cell proliferation, CCK-8 assay was performed on human esophageal carcinoma cell line TE-1 treated with different concentrations of Bortezomib (0, 25, 50, 150, 450, and 1350 nM) for 24, 48, and 72 hours (Figure 1A). A clear increase in cell growth inhibition over time and concentration was observed. The half maximal inhibitory concentration (IC50) values of Bortezomib were 138.4 and 68.03 nM for 48-hour and 72-hour treatments, respectively. A similar effect was also observed in the KYSE-150 cells upon Bortezomib treatment (Figure 1B), although the overall inhibition was less effective. The IC50 values in KYSE-150 cells were 285.1 and 238.2 nM for the 48-hour and 72-hour treatments, respectively. These data indicated that Bortezomib could significantly inhibit the growth of human esophageal carcinoma cells in a dose- and time-dependent manner.

Bortezomib inhibits the proliferation of esophageal carcinoma cells. TE-1 cells (A) and KYSE-150 cells (B) were incubated with Bortezomib at the concentrations (nM) and time (hours) as indicated. The cell viability was assessed by CCK-8 assay and presented as means (SD) from 3 independent experiments (
Bortezomib Causes Cell Cycle Arrest and Apoptosis in Esophageal Carcinoma Cells
In order to investigate how the antiproliferative effect of Bortezomib was mediated, we first analyzed the cell cycle distribution. Although TE-1 cells were treated with increasing doses of Bortezomib (0, 50, 150, 450 nM), G2/M arrest was only observed with the highest concentration (450 nM; Figure 2A). In contrast, KYSE-150 cells started to display G2/M arrest at a much lower concentration of 150 nM (P = .019; Figure 2B). Consist with this, Western blotting analysis showed an induction of cyclin B1 level in both TE-1 and KYSE-150 cells after 24 hours of Bortezomib treatment (Figure 2E and F).

Bortezomib alters the cell cycle distribution of esophageal carcinoma cells. TE-1 cells (A) and KYSE-150 cells (B) were preincubated with Bortezomib at indicated concentrations for 24 hours before being harvested. Cell cycle distribution was assessed by flow cytometry after staining with PI. The percentages of cells in G0/G1, S, and G2/M phase were calculated and presented as means (SD) from 3 independent experiments (
Next, we determined whether Bortezomib slowed down the cell growth via apoptosis induction. As seen with Annexin V-PI staining, increasing doses of Bortezomib severely induced apoptosis in TE-1 cells after 24 hours (Figure 3A). Apoptosis was further enhanced after 48 hours of Bortezomib treatment (Figure 3B). In comparison, the apoptotic population in the KYSE-150 cells only increased significantly after 48 hours of Bortezomib treatment (Figure 3D) but not after 24 hours of treatment (Figure 3C). Consist with this, Western blotting analysis showed an enhanced level of cleaved caspase-3 in both TE-1 and KYSE-150 cells after 48 hours of Bortezomib treatment (Figure 3E and F). These results indicated that Bortezomib caused cell cycle arrest and apoptosis in esophageal carcinoma cells.

Bortezomib enhances the apoptosis of esophageal carcinoma cells. Indicated concentrations of Bortezomib were applied to treat TE-1 cells for 24 hours (A) or 48 hours (B) and KYSE-150 cells for 24 hours (C) or 48 hours (D) before being harvested. Apoptosis was analyzed with FITC Annexin V-PI staining. The percentages of apoptotic cells were presented as means (SD) from 3 independent experiments (
Bortezomib Alters Expression of Genes Involved in Multiple Signaling Pathways
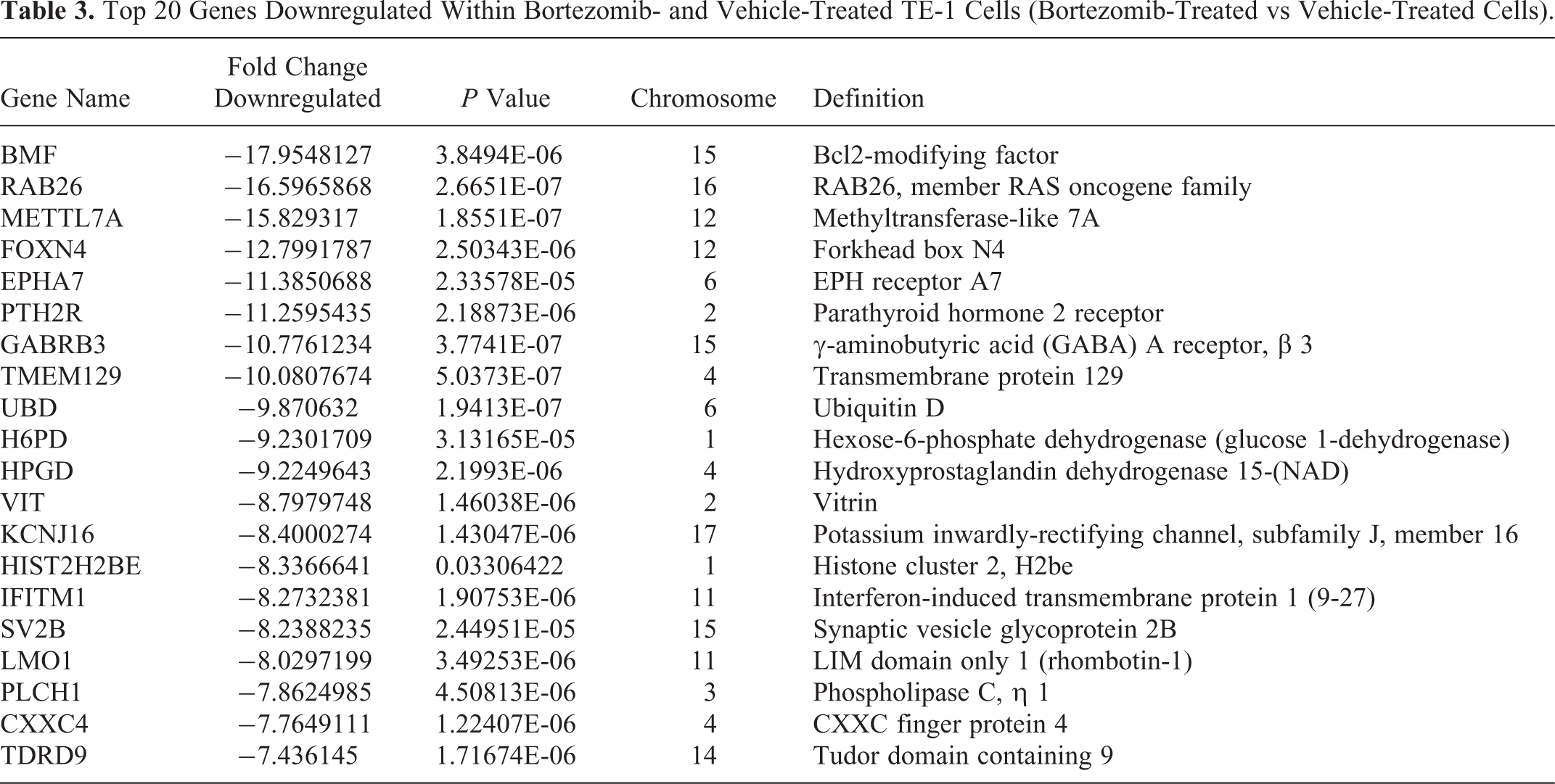
To explore the underlying mechanisms responsible for Bortezomib-mediated cytotoxicity, we profiled genes that were differentially expressed in TE-1 cells in response to Bortezomib. Total RNA was extracted from cells 24 hours after the treatment and microarrayed for more than 44 000 transcript assay probes. The results showed a total of 2975 differentially expressed genes (P < .05) between Bortezomib- and vehicle-treated cells. Among them, 1147 genes were upregulated, and 1828 genes were downregulated (Figure 4A). The top 20 genes dysregulated in the Bortezomib-treated cells in comparison to the vehicle-treated cells are shown in Figure 4B and Tables 2 and 3. Pathway analysis revealed that multiple signaling pathways were significantly influenced via Bortezomib treatment (Figure 4C), such as proteasome, ER, Wnt signaling, calcium signaling, and so on. The expression patterns of 4 representative genes, including 1 upregulated gene GADD45A and 3 downregulated genes UBD, CUL3, and HDAC6, were verified by quantitative real-time PCR (qRT-PCR) and showed consistency with the microarray assay (Figure 4D).
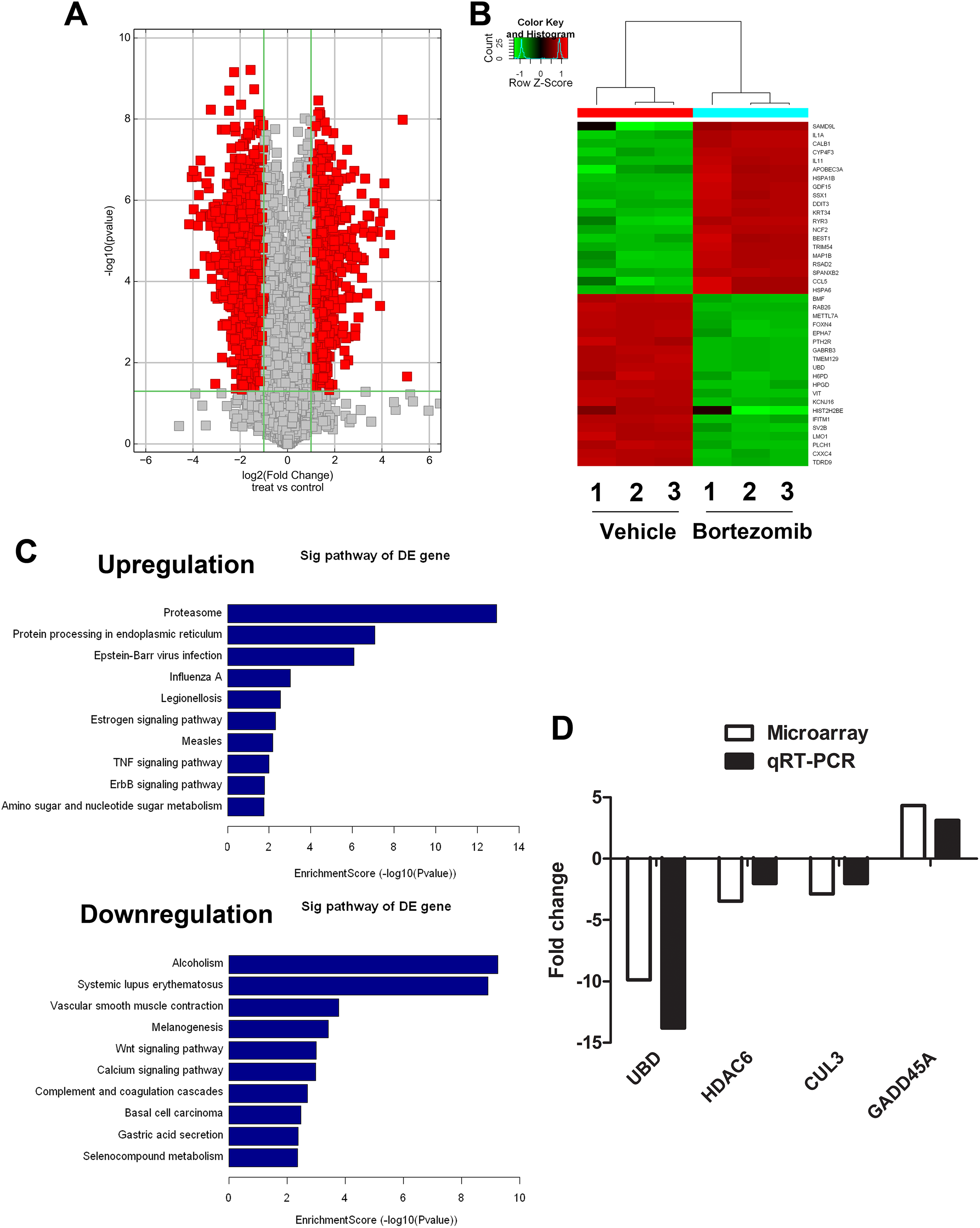
Gene profile analysis in esophageal carcinoma cells in response to Bortezomib treatment. A, Volcano plot comparing vehicle treatment versus Bortezomib treatment. B, Clustering map of top 20 genes dysregulated within Bortezomib and vehicle treated TE-1 cells. C, Significant pathways altered in TE-1 cells (Bortezomib vs vehicle treated cells). D, Quantitative real-time PCR validation for selected genes in Bortezomib and vehicle treated TE-1 cells. PCR indicates polymerase chain reaction.
Top 20 Genes Upregulated Within Bortezomib- and Vehicle-Treated TE-1 Cells (Bortezomib-Treated vs Vehicle-Treated Cells).

Abbreviation: mRNA, messenger RNA.
Top 20 Genes Downregulated Within Bortezomib- and Vehicle-Treated TE-1 Cells (Bortezomib-Treated vs Vehicle-Treated Cells).
Discussion
There is no doubt that numerous studies have affirmed the critical role of ubiquitin–proteasome system in cellular functions and survival. The proteasome has been validated as a novel as well as a valuable molecular target in cancer treatment. As the first proven PI in clinical use, Bortezomib has been widely used to treat various hematological malignancies. Mechanistically, Bortezomib is able to inhibit the activity of proteasome via covalent binding to threonine of the proteasome subunit, 15 therefore, dysregulating the degradation of the proteins involved in multiple signaling pathways as well as inducing apoptosis. 16 The role of Bortezomib in treating solid tumors, however, requires further characterization.
In this study, we observed significant reduction in the proliferation of 2 types of human esophageal carcinoma cell lines, TE-1 and KYSE-150, in a concentration- and time-dependent manner (Figure 1). We also found that in both cell lines, cell cycle was clearly arrested in G2/M at a dose of 150 or 450 nM (P < .05; Figure 2), which is consistent with a previous report. 10,17 Interestingly, 24-hour Bortezomib treatment was sufficient to induce apoptosis in TE-1 cells (Figure 3), while the KYSE-150 cells needed a 48-hour treatment to undergo proper apoptosis. Since TE-1 cells are well-differentiated esophageal carcinoma cells and KYSE-150 cells are poorly differentiated, it is possible that the apoptotic effect of Bortezomib may depend on the degree of tumor differentiation; however, further thorough investigation is required.
The microarray data (Figure 4) suggest that Bortezomib is able to influence multiple signaling pathways. As one of the main modulators in cell death, ER plays an important role in receiving and enlarging the apoptotic signals. 18 Once protein synthesis is initiated, the entire machinery is translocated into ER for further translating and folding. When the proteasomes are inhibited by Bortezomib, the misfolded or redundant proteins accumulate in the ER, which leads to the ER stress and activates caspase-12 for apoptosis. Ca2+ is an important second messenger associated with the regulation of cell proliferation and apoptosis. It is reported that thapsigargin, a Ca2+-ATPase inhibitor, is able to deplete ER Ca2+ stores and induce apoptosis via Bax/Bak. 19 Bortezomib may also induce apoptosis by modulating the intracellular Ca2+ stores.
A number of studies have shown that various mechanisms are possibly involved in Bortezomib-induced growth inhibition and apoptosis, for example, the upregulation of Noxa and repression of Mcl-1 in mature T-cell lymphoma cells, 20 the activation of nuclear factor-κB in MM cells, 21 the reduction of Bcl-2 family proteins in Jurkat lymphoma cells, 22 and so on. Our microarray data further revealed a series of novel potential targets. GADD45A, which plays an important role in genomic stability and DNA damage response, is able to suppress cell growth by inhibiting activity of Cdc2 kinase as well as induce apoptosis by releasing Bim from microtubule-associated components and translocating it to the mitochondria. 23 HDAC6 is a member of the class IIb HDAC family, whose overexpression could accelerate the mobility, migration, and invasiveness of tumor cells. 24 HDAC6 inhibitors exhibit antiproliferative and proapoptotic activities. UBD (also known as FAT10), a ubiquitin-like protein family member, is capable of either mediating the degradation of its target substrates or competing against ubiquitination to stabilize its target substrates depending on the circumstances. Overexpression of UBD has been reported to enhance tumor cell growth by upregulating eEF1A1. 25 Therefore, the upregulation of GADD45A and the downregulation of HDAC6 and UBD indicated by the microarray data and confirmed by qRT-PCR may point toward a mechanism of Bortezomib-induced G2/M arrest and apoptosis. Furthermore, as a member of cullin-RING ubiquitin E3 ligase family, CUL3 is very little understood in tumors, which may also provide a novel target for Bortezomib in cancer therapy.
Footnotes
Authors’ Note
Nannan Ao, MM, and Yingchu Dai, BM were equal contributors. Microarray data are available on the GEO database: accession number GSE97558.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by the National Natural Science Foundation of China (31300694, 81673091), China Postdoctoral Science Fund Program (2015M580463), and Natural Science Foundation of Jiangsu Province (BK20130336). This work was also supported by the Priority Academic Program Development of Jiangsu Higher Education Institutions (PAPD) and Jiangsu Provincial Key Laboratory of Radiation Medicine and Protection.