
Abstract
Since “Warburg effect” has been firstly uncovered in cancer cells in 1956, mounting evidence has supported the molecular mechanism underlying the energy metabolism in induced chemoresistance in cancers. MicroRNAs can mediate fine-tuning of genes in physiological process. MicroRNAs’ energy metabolic role in chemoresistance has been probed recently. In this review, we summarize 5 microRNAs in regulating glucose and lipid metabolism and other energy metabolism. They partially modulate chemoresistance to cancer treatments. Furthermore, we discuss the great therapeutic potential of metabolism-related microRNAs in novel combinatorial means to treat human cancers.
Introduction
In the last decade, reprogramming of energy metabolism has been added to the list of hallmark traits in neoplastic diseases. 1 Arguably, the best characteristic of these metabolic changes is the “Warburg effect.” The Warburg effect is referred to as the process where cancer cells preferentially metabolize glucose through aerobic glycolysis rather than oxidative phosphorylation (OXPHOS). 2 This metabolic switch facilitates biosynthesis of macromolecules and fuels proliferation and invasion of cancer cells. 3,4 In addition, energy metabolic alterations in fatty acid oxidation (FAO) and glutamine also impact tumor growth to certain extent. 5,6 Chemoresistance is recognized as the primary cause of failing chemotherapeutic treatments in human cancers. MicroRNAs (miRNAs) are important regulators in multistep process of animal physiology and diseases including drug resistance. It is reported that dysregulations of specific miRNAs link to chemoresistance via drug efflux increase, alterations in drug targets, DNA repair pathways, evasion of apoptosis, cell cycle control, and so on. 7 -9 Gradually, roles of miRNAs in the association between energy metabolism and chemoresistance have garnered much interest in the field of cancer researches in recent years. In this review, we focus on the miRNAs that modulate chemosensitivity by regulating the energy metabolism of tumors which provide a potential therapy to reverse chemoresistance less or more.
miR-122
miR-122, an abundant liver-specific miRNA, demonstrated the antitumorigenic functions in hepatocellular carcinomas (HCCs). 10 Recent studies have elucidated the pivotal role of miR-122 in conventional chemotherapeutic resistance involved in glucose metabolism. Overexpression of miR-122 resensitizes 5-fluorouracil (5-FU)-resistant colon cancer cells to 5-FU through the inhibition of M2 splice isoform of pyruvate kinase (PKM2) in vitro and in vivo. 11 Besides, elevated miR-122 in HCC overcomes doxorubicin (DOX) resistance through decreasing PKM2, without influencing the intracellular DOX accumulation. 12 Pyruvate kinase regulates the final rate-limiting step of glycolysis accompanied by dephosphorylation of phosphoenolpyruvate to pyruvate. It’s reported that downregulation of PKM2 expression is able to reverse gemcitabine resistance in pancreatic cancer cells. 13 But intriguingly, PKM2 enhances chemosensitivity to cisplatin through interaction with the mammalian target of rapamycin (mTOR) pathway in cervical cancer. 14 In addition to miR-122, other miRNAs could also regulate PKM2 expression in human cancers, such as miR-133a and miR-133b in tongue squamous cell carcinoma, 15 miR-326 in glioblastoma, 16 and miR-338-3p in ovarian cancer. 17 But further investigations are needed to elucidate the mechanisms underlying these miRNAs in metabolic regulations and their potential to modulate chemoresistance.
Meanwhile, miR-122 also shows pleiotropic effects on chemoresistance in cancers via other metabolic reprogramming phenotypes. Solute carrier family 7 (SLC7A1) was identified as an arginine transport as well as the direct target of miR-122. In HCC, silenced miR-122 expression causes upregulation of SLC7A1 and increased intracellular arginine levels, which render sorafenib-resistant cancer cells. 18 Furthermore, miR-122 regulates cholesterol and fatty acid metabolism. Systemic miR-122 inhibition by antisense oligonucleotide leads to a broad reduction in plasma cholesterol by regulating the genes involved in cholesterol biosynthesis. 19 Esau et al discovered that miR-122 inhibition led to reduced serum fatty acid synthesis and increased hepatic FAO by causing effect on several lipogenic genes, including reduction in stearoyl-coA desaturase-1 (SCD-1), a rate-limiting enzyme in lipogenesis and acetyl-CoA carboxylase 1 (ACC1), regulator of malonyl-CoA production. 20 This is of significance because fatty acids and cholesterol can serve as a kind of energy storage and components for biosynthesis of cellular biomembrane which aid tumor cells in maintaining high rate of proliferation. However, the underlying mechanisms between miR-122 regulation and chemosensitivity in human cancers involved in fatty acids and cholesterol synthesis remain active investigations.
miR-125b
miR-125b has been considered as a diverse factor in anticarcinogenic therapy depending on the cellular contexts. Ectopic expression of miR-125b reverses DOX resistance in human breast cancer. 21 But inversely, miR-125b could desensitize Ewing sarcoma cells to DOX by suppressing p53 and Bak. 22 miR-125b overexpression was previously verified in modulating glucose, glutathione, lipid, and glycerolipid metabolism, with implications for inhibition of metabolic adaption of B cells to their malignant phenotype. 23 We further dissect the potential function of miR-125b in the resistance to chemotherapy. There was also an indication of downregulated level of miR-125b in 5-FU-resistant HCC cells. miR-125b is able to inhibit glucose metabolism by directly targeting hexokinase 2 (HK2). Consequently, it performs tumor-suppressor function to reverse 5-FU resistance in HCC cells. 24 In chondrosarcoma cells, overexpression of miR-125 can enhance the sensitivity of both parental and DOX resistant cells to DOX through direct targeting on the ErbB2-mediated upregulation of glycolysis. 25 Hexokinase 2 catalyzes the first irreversible step in aerobic glycolysis by which glucose is phosphorylated into glucose-6-phosphate. A retrospective study showed that HK2 overexpression was associated with chemoresistance and was an independent prognostic indicator for early tumor recurrence in epithelial ovarian cancer. 26 What’ s more, blocking SLUG/HK2 pathway by curcumin noteworthily reversed 4-hydroxytamoxifen resistance in breast cancer cells. 27 Data imply that aim at HK2 targeted by miR-125b directly may serve as an attractive approach to render cancer cells more sensitive to chemotherapeutic reagents.
In maintaining high rates of growth and proliferation inside tumor cells, especially in chemoresistant cancers, nucleotide, membrane phospholipid, fatty acid, and other kinds of macromolecules are predominantly acquired. We further explore the effect of miR-125b on the underlying drug resistance mechanisms implicated in the above multiple processes. In primary adipocytes, miR-125b mimic can decrease lipid droplets and triglyceride (TG) accumulation versus the control by directly regulating SCD-1. 28 Estrogen strongly boosts miR-125b level via ERα in HepG2 cells. Ovariectomized or liver-specific ERα knockdown mice treated with miR-125b-overexpressing adenoviruses were resistant to hepatic steatosis, due to decreased fatty acid uptake and decreased TG synthesis. 29 However, the potential behavior of miR-125b implicated in the aforementioned energy reprogramming in chemoresistance needs more investigations. Moreover, given that miR-125b exhibits dual function in regulating chemosensitivity, 21,22 it remains an energy metabolic topic for intensive and exciting researches in chemoresistant cancer cells.
miR-34a
miR-34a acts as a tumor suppressor in multiple solid tumors. It can antagonize several oncogenic processes including cell proliferation, apoptosis, stemness, metastasis, and drug resistance by modulating various cellular signaling pathways. 30 Although extensive evidence has uncovered the antichemoresistant mechanisms of miR-34a in different cellular pathways, its potential role in energy reprogramming was not addressed until recently. Elevated miR-34a expression reduces level and activity of lactate dehydrogenase A (LDHA) in 5-FU-resistant colon cancer cells, which resensitizes colon cancer cells to 5-FU. 31 Additionally, overexpression of miR-34a renders radioresistant HepG2 cells sensitivity to radiation through the inhibition of LDHA. 32 Lactate dehydrogenase A, one of the predominant isoforms of LDH, is responsible for the conversion of pyruvate to lactate in the last cellular glycolytic reaction. 33 It has been revealed that inhibition of LDHA resensitizes breast cancer cells to Taxol 34 and chondrosarcoma cells to DOX. 35 Collectively, it is cued that targeting miR-34a-LDHA axis could modulate chemosensitivity in diverse resistance types of cancers. p53-inducible miRNA-34a could regulate glucose metabolism via repressing glycolytic enzymes (HK1, HK2, glucose-6-phosphate isomerase [GPI]) and pyruvate dehydrogenase kinase 1 (PKD1) in HCT116 (wild-type p53) cells. 36 But the functional cancer relationship between miR-34a and its downstream targets HK1, HK2, GPI, and PKD1 in chemosensitivity remains unclear.
Accumulating evidence has elucidated that miR-34a overexpression and its downstream targets sirtuin 1 (SIRT1) downexpression attenuate the chemoresistance in human colon cancer and prostate cancer. 37,38 We try to address the potential association between miR-34a-SIRT1 axis and other energy metabolic phenotypes. Sirtuin 1, as a key mediator of beneficial effects of caloric restriction, regulates lipid and glucose metabolism in an NAD+-dependent manner by deacetylating. 39 It has been proposed that a farnesoid X receptor (FXR)-miR-34a-SIRT1 regulatory loop plays a causative role in the pathological process of hepatic lipid metabolism in livers. Activation of FXR signaling smartly decreased miR-34a expression, subsequently resulting in increase in hepatic SIRT1 levels. In metabolic diseases, elevated miR-34a suppresses the expression of SIRT1 and further decreases FXR activity, which delineates a vicious FXR/miR-34a/SIRT1 circuit. 40,41 Likewise, isocaloric pair-fed high-carbohydrate diet induced greater severity in hepatic steatosis and inflammatory response and cholesterol deposition than high-fat carbohydrate diet in mice potentially through higher expression of hepatic miR-34a and lower levels of SIRT1 axis. In detail, this faulty axis elevated inflammatory cytokine genes (Il1β, Tnfα, and Mcp1) to induce hepatic inflammation, increased de novo lipogenesis through related proteins expression (ACC, SCD1), suppressed FAO via downregulating relevant genes (Cpt1, Pparα, and Pgc1α), and decreased expression of cholesterol metabolism-related genes (Abcg5, Abcg11, and Cyp7a1 etc). 42 Apart from miR-34a/SIRT1 axis, miR-34a is highly induced in human liver tissues with nonalcoholic steatohepatitis, diabetic mice, and high-fat diet-fed mice models and its expression inversely correlates with hepatocyte nuclear factor 4α, which leads to very-low-density lipoprotein (VLDL) secretion inhibition in hepatocyte and diminished levels of low-density lipoprotein, VLDL, and TG in plasma. 43 Although the role of miR-34a in chemoresistance in aforementioned metabolic reprogramming remains unclear, it is still significant because if delivery of miR-34a is applied into personalized cancer therapy to cancel the chemoresistance, 37,38 it is critical to take comprehensive administration to attenuate side effects in lipid and cholesterol metabolism.
miR-155
miR-155 functions as an oncomiR and is significantly elevated in human cancers. 44 Inhibition of miR-155 can restore sensitivity of lung cancer to cisplatin via negative regulation of Apaf-1 expression 45 and reverse tamoxifen resistance by activating suppressor of cytokine signaling 6-signal transducer and activator of transcription 3 pathway in breast cancer. 46 However, miR-155 also exerts a possible tumor-suppression function to attenuate cisplatin resistance in human epidermoid carcinoma cells. 47
A recent study has demonstrated that miR-155 drives metabolic reprogramming in estrogen receptor–positive (ER+) breast cancer cells following long-term estrogen deprivation (LTED). 48 In this study, aerobic glycolysis is enhanced through higher expressions of GLUT1 (glucose importer), HK2, lactate exporter monocarboxylate transporter-4 in MCF7-LTED cells, and an aromatase inhibitors (AIs)-resistant breast cancer line. Yet, miR-155/HK2 axis was further deciphered to be associated with the response to AI therapy and tumor plasticity in ER+ breast cancers. Here, miR-155 augment and miR-143 reduction in MCF7-LTED cells are consistent with Jiang and his colleagues’ study that miR-155 promotes HK2 transcription by activation of STAT3 and suppression of miR-143. 49 In other metabolic phenotypes, overexpression of miR-155 reduced total cholesterol, TG, and high-density lipoprotein, and free fatty acid both in serum and hepatic cells via lower carboxylesterase 3/triacylglycerol hydrolase in Rm155LG/Alb-Cre double transgenic mice. 50 Collectively, it’s smoothly reasoned that miR-155 might be a potent metabolic regulator that mediates response to chemotherapy in malignant tumors.
miR-205
As same as miR-125b, miR-205 exhibits dual properties involved in chemosensitivity in different cancer types depending on the specific tumor contexts. 51 -53 Overexpression of miR-205 promoted the growth, metastasis, and chemoresistance of non-small cell lung cancer by targeting phosphatase and tensin homolog deleted on chromosome 10 (PTEN). 52 However, overexpression of miR-205 is able to rescue melanoma cells from drug resistance by reduced levels of Bcl-2, ABCA2, and ABCA5 (ABC transporter family members). 51 More evidence delineates the association with the role of miR-205 in drug-induced metabolic adaptations and chemoresistance in cancers recently. Docetaxel-resistant PC3 cells can utilize glucose, glutamine, and lactate by the OXPHOS; therefore, it has more efficient respiratory phenotype than sensitive cells. Here, reexpression of miR-205 is able to shift OXPHOS to a glycolysis metabolism, thereby resulting in restoral of chemosensitivity to docetaxel. 54
Conclusions and Future Directions
Mechanistic studies were conducted to discuss the roles of 5 miRNAs and their targets in energy metabolic regulation in human cancers, especially in chemoresistance (see Table 1 and Figure 1). Given a specific miRNA could bind to 3′-untranslated regions of different targeted messenger RNAs, fine miRNA mediations and the synergy effects of multiple miRNAs may skillfully alter energy metabolism to adapt tumor chemoresistant condition in human cancer cells. It is promising that liposomal formulation of miR-34 (MRX34) was the first miRNA-based antineoplastic agent to enter phase I clinical trial for advanced HCC or other solid tumors on April 2013 (https://clinicaltrials.gov/show/NCT01829971). Full usage of novel technologies, such as miRNAs mimics and inhibitors, can potentially normalize the dysregulated energy metabolic enzymes and signaling pathways in chemoresistant cancer cells. Moreover, given that miRNAs regulate target genes involved in multiple cellular bioprocesses, they can function as oncogenes or tumor suppressors in different tumor entities. These miRNAs, can, thereby, act as brakes or triggers on the progression of chemotherapeutic resistance in human cancer cells. The insights highlight that miRNAs may serve as novel candidates for therapeutic intervention to elevate therapeutic efficiency for human cancers in the future.
The 5 miRNAs in Energy Metabolic Changes.
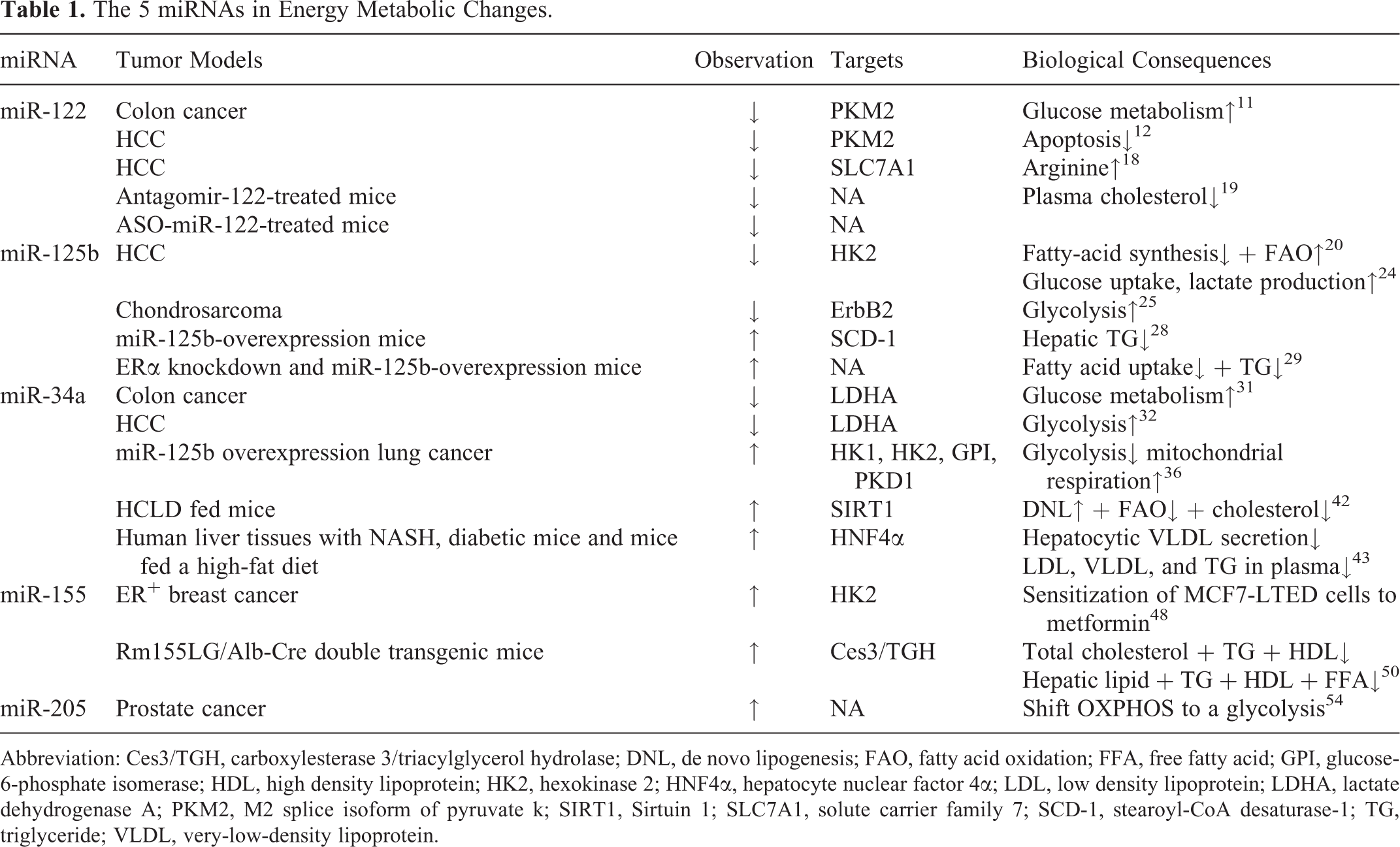
Abbreviation: Ces3/TGH, carboxylesterase 3/triacylglycerol hydrolase; DNL, de novo lipogenesis; FAO, fatty acid oxidation; FFA, free fatty acid; GPI, glucose-6-phosphate isomerase; HDL, high density lipoprotein; HK2, hexokinase 2; HNF4α, hepatocyte nuclear factor 4α; LDL, low density lipoprotein; LDHA, lactate dehydrogenase A; PKM2, M2 splice isoform of pyruvate k; SIRT1, Sirtuin 1; SLC7A1, solute carrier family 7; SCD-1, stearoyl-CoA desaturase-1; TG, triglyceride; VLDL, very-low-density lipoprotein.

MicroRNAs and their targets in the association between energy metabolism and chemoresistance in cancer cells.
Footnotes
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by funds from the Chinese National Natural Science Foundation No. 81472760S (L. H.), the Guangdong Natural Science Funds No. 2014A030313057 (J. Y.), and the Science and Technology Program of Guangdong No. 2014A020212078(L. H.) and No. 2013B021800088 (J. Y.).