
Abstract
The aim of this work was to develop flame-retarded polyamide 6 fibers using the hexaphenoxycyclotriphosphazene (HPCP) as a flame retardant by melt spinning. The influence of HPCP on the spinning process was investigated. HPCP caused a lower melting point and a higher crystallinity of polyamide 6 chips. The melt spinning temperature of the polyamide 6/HPCP composite fibers was less than the pure polyamide 6 fibers and the achievable draw ratio was determined. Structural and morphological characterization of the melt-spun fibers demonstrated good dispersion of HPCP additive. The introduction of HPCP did not change the crystal configuration of fibers in which the γ-crystal phase was predominant. The loss of mechanical properties of polyamide 6 fibers was mitigated by HPCP. The thermal behavior and flame retardancy of the polyamide 6/HPCP composite fibers was also enhanced effectively.
Introduction
Polyamide 6 (PA6) is one of the polymeric textile materials with wide application in building, home textiles and transportation [1,2]. The most outstanding characteristics of PA6 fibers are the excellent toughness, wear, high impact strength and abrasion resistance [3]. However, the flammability and severe dripping during combustion limit the above applications of PA6 fibers. As a consequence, developing flame-retarded PA6 fibers with less dripping is conforming to the trend of industrial development. Flame-retarded PA6 fibers or fabrics can be achieved in several methods [4,5]: (i) using flame-retarded monomers during synthesis and grafting the molecules on the polymeric chain, (ii) incorporation of flame retardant (FR) additives into polymer before spinning process, (iii) FR finishing applied to the fabrics. The approach of melt blending with FRs before spinning is favored because it is technically easy and efficient [6]. However, the suitable FRs for PA6 in textile remain an ongoing problem because of the PA6 high melt spinning temperature [1] which requires the FRs to have appropriate decomposition temperature. The introduction of FRs to PA6 during spinning caused discontinuity of the melt microflow and easy fracture of single filament in the preparation process. In addition, the high loading of FRs in fibers is needed due to the thin textiles, which adversely affects the spinnability of the PA6 fibers [7]. So far, much work has been focused on flame-retarded PA6 resins [8,9]. Effective FRs, such as phosphorus- or nitrogen-based additives and nano-particles have been developed in recent years to investigate the flame retardancy of PA6 [10,11]. There have been some researches focusing on the preparation of flame-retarded PA6 fibers. Xiang et al. [10] investigated the synergistic effect between α-zirconium phosphate and ammonium sulfamate (AS) for enhanced flame-retardant properties of PA6 fibers. They found that the addition of AS provided negative effects on the tensile strength of PA6/AS/α-ZrP; however, the adverse trends that mentioned above could be overcome by using the well-dispersed α-ZrP. In order to obtain acceptable tensile properties of flame-retarded PA6 fibers, Wu et al. [12] prepared flame-retardant nanocomposite PA6 fibers with nanoclay and intumescent additives by melt spinning. The additives had good dispersibility in the fiber and the mechanical strength was not greatly decreased. However, it is not the only solution to solve the problem. The development of flame-retarded PA6 fibers with comprehensive properties using single component FR needs more investigation.
Cyclophozene consists of alternating phosphorous and nitrogen atoms that are connected with single and double bonds [13]. In recent years, there has been considerable interest in the phosphazene-based materials owing to their excellent thermal and flame-retarded properties to polymers. Hexaphenoxycyclotriphosphazene (HPCP) is known as a phosphonitrilic FR whose molecule structure is shown in Figure 1. It not only avoids the pollution caused by halogen but has outstanding flame retardancy and acceptable thermal stability [14,15]. Li et al. [16] prepared the flame-retarded viscose fiber infused with HPCP, and they found that HPCP benefitted the flame retardancy to viscose fibers and did not impact the mechanical properties. Therefore, we assumed that HPCP can support the melt spinning of PA6 fibers owing to its low melting point and improve the flame-retarded property. It is possible to prepare flame-retarded PA6 fibers with excellent performance through not adding other dispersants but controlling the process parameters due to heterogeneous nucleation. As a result, HPCP is selected as the FR in this study to investigate the spinnability which is defined as the ability of a fluid to form continuous filaments under tension [17]. It is not only related to the quality of the chips but also related to the spinning process. In the previous research, the spinning process of flame-retarded PA6 fibers by melt spinning has not been studied.

Chemical structure of HPCP.
In this study, PA6 fibers with different ratios of HPCP were prepared via melt spinning. The influence of HPCP on the melting and crystallization behavior and apparent viscosity of PA6 chips was investigated. The spinning technology involving spinning temperature and the stretching process of PA6/HPCP fibers was determined. To analyze the performance of the PA6/HPCP fibers, investigations including morphology, crystalline structure, orientation, tensile properties and flame retardancy were performed.
Experimental
Materials
Chips of textile grade PA6 polymer, with a high relative viscosity equal to 3.2, were provided by Jiangsu Hongsheng New Material Co., Ltd. HPCP of commercial grade was purchased from Zibo Lanyin, Co., Ltd. It was in the appearance of tiny particles with diameters in the range of 0.1–0.3 mm. HPCP has a melting point of 140–150°C, a crystallization temperature of 60°C and a decomposition temperature of 330°C. The chips of 10 wt% HPCP in PA6 (PA6/HPCP-10) and 20 wt% HPCP in PA6 (PA6/HPCP-20) were used as masterbatches in combination with pure PA6 chips to obtain composite formulations with different contents of HPCP. The PA6 chips and as-prepared masterbatches were dried at 105°C before use.
Processing of the PA6 composite fibers
The pure PA6 and PA6/HPCP composite fibers were prepared with melt spinning process. The spinneret for multifilament spinning has a diameter of 0.35 mm. PA6 chips were added to the hopper which is the feeding zone of the single screw extruder. In the extruder, the chips and masterbatches with evaluated ratios were mixed and melted. The screw of the extruder pushed the melt to the spinning pack. The polymer melted in the spin pack was filtered through the 35 µm filter in order to remove aggregates. The melt passed into the cooling shaft through the spinneret, where it was solidified and packed onto the winding drum with the velocity of 850 m/min. The extruded formulations under the conditions are detailed in Table 1. The melt spinning process of the PA6/HPCP composite fibers is illustrated in Figure 2.
Composition (wt%) of the different formulations in spinning.
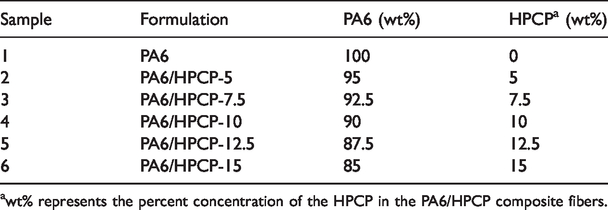
awt% represents the percent concentration of the HPCP in the PA6/HPCP composite fibers.

The melt spinning process of PA6/HPCP fibers.
Characterization of the samples
Measurement of properties of the chips
Differential scanning calorimetry (DSC) was carried out on STA449C thermal analyzer (NETZSCH, Germany) with a flow rate of 20 ml/min under nitrogen. The temperature changed in the range of 30 to 300°C; the heating and cooling rates were set to 10°C/min. Samples were placed in the standard aluminum sample pans to determine their melting temperatures, Tm, the crystallization temperature, Tc, and percentage crystallinity, Xc, respectively. The percentage crystallinity values of the specimens were calculated using the equation below
Apparent viscosity (ηa) of the PA6/HPCP formulations was investigated using RH2000 capillary rheometer (Rosand, England). The diameter of the capillary was 0.5 mm with an L/D (length to diameter) ratio of about 25. The test was performed at the shear stress of 40.5 MPa and temperatures range from 260°C to 295°C with a heating rate of 5°C/min. All samples were dried in a vacuum oven at 80°C for16 h and molded into disks before the test.
The diameter of the fibers was measured by GL002C fiber fineness comprehensive analyzer, each fiber sample was tested 100 times randomly and averaged. The typical images were saved by optical microscopy. The morphology of the fibers was recorded by scanning electron microscopy (SEM) using S4800 II field emission scanning electron microscope (Hitachi, Japan) accelerating voltage of 7 kV. All the samples were sprayed with gold before observation.
X-ray measurements were performed using a TD-3700 X-ray diffractometer, with a voltage of 40 kV and a current of 150 mA, using CuKα (λ = 0.15418 nm) radiation. The powdered fiber samples were scanned from 5° to 40° (2θ) at a rate of 10°/s and a step size of 0.02.
YG001D monofilament tensile testing machine was used for the tensile test of the fiber samples. The drawing speed was 20 mm/min and the gauge length was 20 mm. Each formulation of the fiber samples was tested 20 times. All fibers were conditioned at 25°C and 65% relative humidity before measurement. The orientation factor values (fo) of the fiber samples were measured using an SCY-III sonic velocity orientation instrument. Before testing, the fiber samples were wound and clamped on the device with a span of 20 cm and 40 cm. Each fiber sample was tested five times and the main fo values were obtained [19].
Thermogravimetric (TG) analyses were performed using an STA 449F3 instrument (NETZSCH, Germany) in the entire temperature range of 30 to 800°C at a heating rate of 10°C/min in an air atmosphere.
The limiting oxygen index (LOI) values of the materials were evaluated with an M606B digital oxygen index tester (Qingdao Shanfang, P.R. China) according to the Chinese national standard GB/T5454-1997. The number of molten drops within 10 s was recorded. Three measurements were examined for each fiber sample. The burning behavior of the fibers was conducted with 45-degree slope burning test in Japan industry standard JIS L 1091-1992, using a GT-C32 45-degree flammability tester (GESTER, China). The fiber strand specimens of around 10 cm long, 1 cm wide and 1 cm thick were prepared by twisting of several filaments together. The fibers were heated for 30 s under flame source, then the afterflame time and afterglow time were recorded. The damaged length was also measured. Under the same condition, the ignition times of fibers burning 90 mm were also reported. Three specimens were used for each test.
Results and discussion
Differential scanning calorimeter
DSC was used to analyze the influence of HPCP on the melting and crystallization behavior of PA6. The DSC curves are depicted in Figure 3, and the results which contain the transition temperatures, such as the melting temperature (Tm), the crystallization temperature (Tc), as well as the percentage crystallinity (Xc) are presented in Table 2. It is necessary to analyze the melting property which is basic to set the spinning temperature of PA6/HPCP composite fibers. The spinning temperature is the most important parameter to determine the formation and quality of the fibers in the spinning process. As depicted in Figure 3(A), a decrease in Tm was observed for PA6 fibers with the addition of HPCP, which indicated that HPCP acted as a plasticizer. The lubrication effect in PA6 melt can make the molecular chain easier to move. Therefore, the temperature of each zone of the screw extruder can be lower than the spinning temperature of pure PA6 chips. It is interesting to find that the main endothermic melting peaks of PA6/HPCP-10 at around 224.9°C corresponded to the α-crystal phase, and the shoulder peaks at 190°C corresponded to the γ-crystal phase [20,21]. This confirmed that both α-crystal and γ-crystal phases were existed in the PA6 chips.

DSC heating (A) and cooling (B) curves of PA6 (a), PA6/HPCP-5 (b), PA6/HPCP-7.5 (c), PA6/HPCP-10 (d), PA6/HPCP-12.5 (e) and PA6/HPCP-15 (f).
The melting temperature, Tm, the crystallization temperature, Tc, the initial crystallization temperature, Tci, the enthalpy of melting, ΔHm, and the degree of crystallinity, X c , of the pure PA6 and composite PA6/HPCP chips.

As shown from the DSC cooling curves in Figure 3(B), Xc values increased from 5.88% to 7.81%. The values of initial crystallization temperature (Tci) and Tc of PA6/HPCP chips consistently increased with the increase in their HPCP contents. Apparently, even minor contents of HPCP can serve as efficient nucleation sites for PA6 molecules during their crystallization processes. HPCP played a role of heterogeneous nucleating agent during the crystallization process, resulting in the chain segment crystallization in PA6 non-crystallization zone and an increase of crystallization rate. Therefore, the cooling speed should be appropriately reduced by decreasing quench flow rate, which is conducive to improve the strength and evenness of the fiber.
Spinning technology of the PA6/HPCP fibers
In the process of melt spinning, the high spinning temperature can intensify the thermal decomposition of polymers and accelerate the movement of macromolecules in the fiber [22]. The macromolecules are less prone to orientation due to the low melt viscosity at high temperatures.
The apparent viscosity test of the PA6 formulations was carried out to evaluate the impact of the HPCP on melt spinning performance. The apparent viscosity (ηa) of pure PA6 and PA6/HPCP formulations is plotted in Figure 4. Neat PA6 has the highest melt viscosity compared to its composite formulations. HPCP leaded to a decrease of the ηa, indicating the higher flowability in PA6/HPCP composites. HPCP acted as a plasticizer in PA6 and advanced the flow of composite melts. Because of such significant decrease in viscosity, samples with higher HPCP loadings were suitable for fiber spinning due to good spinnability. For all formulation, the ηa declined with temperature heating but varied slowly when the temperature was above 275°C. Combined with the DSC curves, the decreased melting point determined that the melt spinning temperature and cooling rate of the composite fibers can be lower than the common PA6 fibers.

Melt viscosity (ηa) at different temperatures and HPCP content.
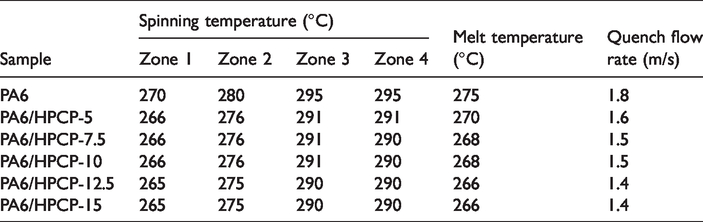
All-spinning zone temperatures of the extruder with stable spinning production and quench flow rate are presented in Table 3. Based on the repeated experiments, the melt spinning temperature was selected to be 4–5°C lower than pure PA6, which can remain in available viscosity to provide the melt to present uniform and continuous state as well as stable melt pressure. It is expected that the PA6/HPCP system should be spinnable at lower than 20 wt% loading. However, we find that fusant was difficult to form continuous melt flow and the spinneret was blocked when the content of HPCP was above 15 wt%. This may be because the higher content of HPCP dispersed poorly in the matrix and the melt mass was uneven. Therefore, it can be concluded that the HPCP can be applied to spin PA6 fibers with good spinnability at a reasonable range of content.
All-spinning zone temperatures of the extruder.
After extruded, the melt was drawn into filaments due to the action of a tensile force. Certain typical orientation factor (fo) values and mechanical properties at varying draw ratios are summarized in Table 4. The diameters decreased slightly with the increase in the draw ratios. The fo value was presented as a maximum value with a draw ratio at 4.6 for PA6/HPCP-10 fibers in this study. Contrary to expectation, the breaking tenacity and elongation at break increased first and then decreased with the increase in the draw ratio. This may be explained by the fact that the higher draw ratio may cause the macromolecules inside the fibers to slip and break, leading to a decrease in the mechanical properties. At this draw ratio, the fo values remained at relatively high values, indicating the neat axial arrangement of the molecular chain and the compact internal structure of the fibers. As a consequence, other achievable PA6 fibers with acceptable mechanical property were produced at a draw ratio of 4.6.
The orientation factor (fo) values and tensile properties of PA6/HPCP-10 fibers at different draw ratios.

The dispersion and diameters of the PA6/HPCP fibers
Figure 5 presents the optical microscope photographs showing the diameters of PA6 and PA6/HPCP formulations. Fiber diameter analysis results are shown in Figure 6. The diameters of all the PA6 fibers were found to be around 35 µm, though there was a trend to become fine in diameters. It can be explained by that the incorporation of HPCP reduced the polymer viscosity which is related to the polymer throughput during extrusion. It is in agreement with the literature that the extrusion temperature and the quench flow rate can cause mild changes in diameters [23].

Optical microscope photographs of PA6 fibers. (a) and PA6/HPCP-10 (b) fibers.

Fiber diameter analysis results.
The SEM images of the fiber cross-section were used to investigate the dispersion of the HPCP. There were fine particles in the cross-section of PA6/HPCP fibers compared with the smooth cross-section of pure PA6 fiber, as revealed in Figure 7. HPCP dispersed uniformly in the fiber, without obvious agglomeration. This was because HPCP can melt firstly due to the low melting point and then dispersed into PA6 to get uniform dispersion, an important feature that can remain as the mechanical property of the fibers.

SEM images of the cross-section of PA6 fibers.
X-ray diffraction
X-ray diffraction (XRD) was utilized to study the effect of HPCP on the crystal configuration and crystallinity of the spun PA6 fibers. From Figure 8, it can be seen that all the PA6 fiber samples had distinct diffraction centered peaks at 2θ = 10.8° and 21.7° corresponding to γ-crystal phase [7]. It indicated that the γ-crystal phase of PA6 was predominant in fibrous samples, though the α-crystal phase was more stable. This can be concluded that the fibers were favored to γ-crystal phase due to the lower cooling rate [12]. The incorporation of HPCP did not change the crystal configuration of PA6 fibers. It is interesting to find that the intensity of diffraction peak decreased gradually with the increase of HPCP when the content was within 10 wt%, which means that the crystallinity of PA6 fibers decreases with the increase of HPCP. The crystallinity was slightly increased when the content of HPCP was above 10 wt%. This stems from the fact that the amounts of γ-crystal phase of fibers increased with higher content of HPCP, which plays a role of heterogeneous nucleating agent and enhanced the crystallinity [24].

XRD patterns of pure PA6 and PA6/HPCP fibers.
Mechanical properties
Tensile test and orientation factor (fo) values were made to study the potential effects of the HPCP particles on the mechanical properties, which can indirectly evaluate the spinning process. The test results of different PA6 fibers are presented in Figure 9, and stress–strain curves are shown in Figure 10. It can be seen that the breaking tenacity slightly decreased as the HPCP increases. With the addition of 10 wt% and 15 wt% HPCP, the breaking tenacity of PA6 fibers decreased from 5.63 to 4.86 and 3.64 cN/dtex, namely by 14% and 35%, respectively. This is consistent with the SEM image of the cross-section of the PA6/HPCP fibers, indicating HPCP at 10 wt% content had better dispersion than that at 15 wt%. The higher amount of HPCP can slide against each other and serve as the defects during the tensile processes of PA6/HPCP fibers, leading to a reduction in their breaking tenacity consequently. However, the changes were not significant, and HPCP mitigated the potential loss of breaking tenacity. Meanwhile, the fo value showed a downtrend trend with the increase in HPCP content. At the 15 wt% HPCP loading, the fo value of PA6 fibers showed a moderate reduction. As evidenced by XRD analyses in the previous section, the major γ form of PA6 crystal was obtained from melt spinning. However, more perfect γ form crystals can be well arrayed as HPCP contents enhancing. On the other hand, HPCP caused higher elongation at break, a feature that represents the flexibility of the fibers. The elongation at break can reach approximately 143.52% at the 10 wt% loading of HPCP. This may be due to the plastic property of HPCP. From Figure 10, there is no necking stage for the fibers due to the slow cooling rate and the same effect has been also found in the research of other FRs [12].
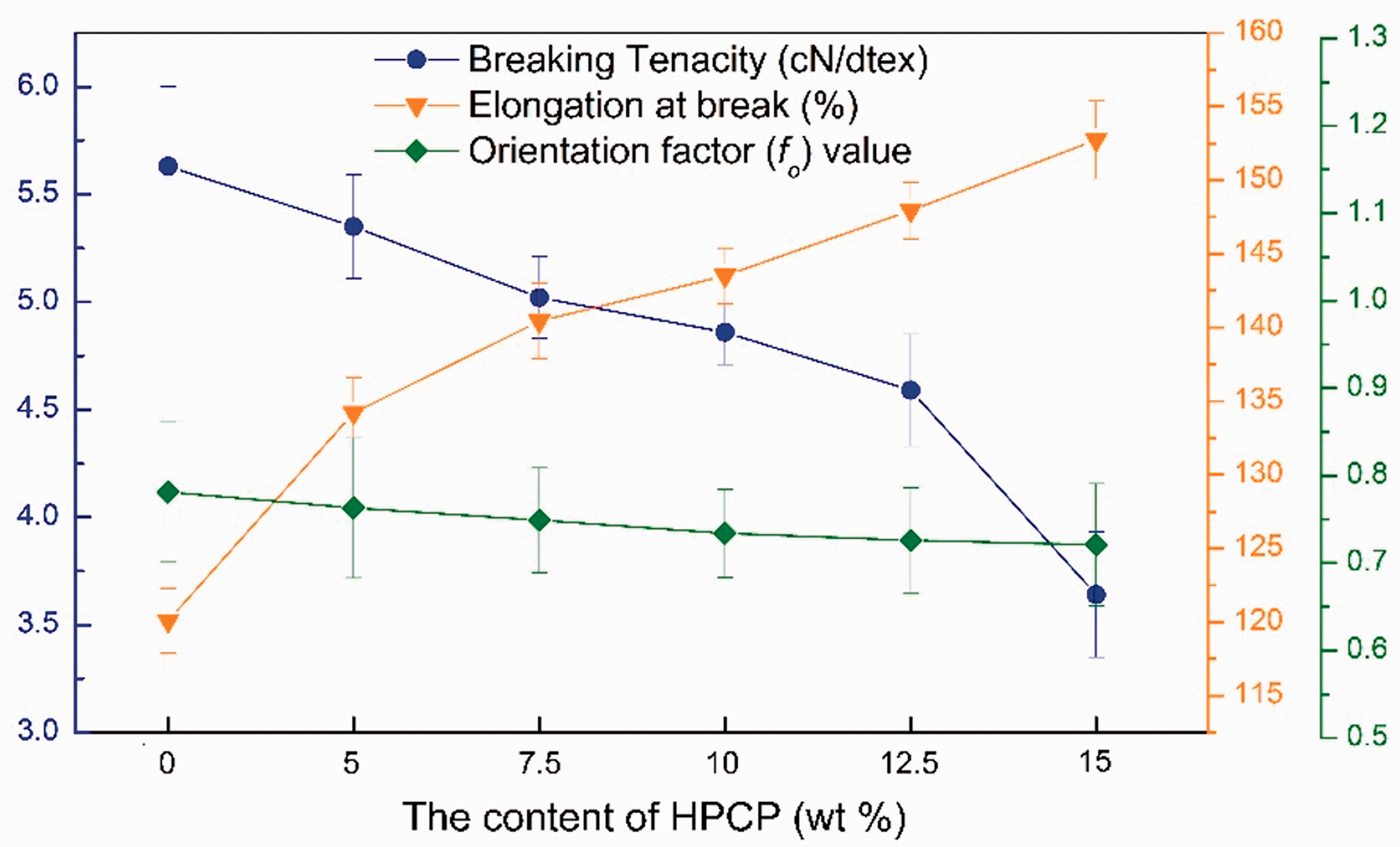
Breaking tenacity and elongation at break of pure PA6 and PA6/HPCP fibers.

Typical stress–strain curve of PA6 and PA6/HPCP-15 fibers.
Thermal behavior
Figure 11(a) and (b) depicts the TG and DTG curves of the pure PA6 and PA6/HPCP-10 fibers. The thermal degradation can be seen as two stages. The first decomposition stage was derived from the decomposition of the composite and the formation of the char. The second stage was that the char would be reacted again due to the thermal instability in air [25]. In the process of PA6/HPCP, the first degradation was starting at 388.05 °C. At this stage, HPCP was degraded and produced phosphoric acid and metaphosphoric acid, and then it would generate polymetaphosphoric acid, which would accelerate the dehydration of the materials and form phosphorous oxide [26,27]. The introduction of HPCP caused that the initial degradation temperature of the composites and the maximum decomposition rate temperature decreased to about 20°C. The second stage began at 503.26°C. The residual mass at 800°C of the composites increased from 0.23% to 1.69%, which identified that the HPCP can promote the carbonization of PA6 and enhance the char stability.

The TG and DTG curves of PA6 (a) and PA6/HPCP-10 (b) fibers.
Flame retardancy of fibers
The limiting oxygen index test was to evaluate the flame retardancy of the fiber samples. The results of the LOI value are shown in Figure 12. The LOI value of PA6/HPCP fibers increased from 23.4 to 29.8 with the increase of HPCP, which was up to the flame-retarded standard [28]. The incorporation of HPCP reduced melt dripping significantly. The improvement of the flame retardancy could be explained by the fact that HPCP had satisfactory thermostability because of the alternating ring structure of phosphorus and nitrogen atoms which exhibit the synergistic flame-retarded effect. Meanwhile, the decomposition products of phosphazene groups during combustion promoted the charring of PA6 fibers [29]. The formed char layer inhibited the air contacting with the materials and reduced the efficiency of heat transfers.
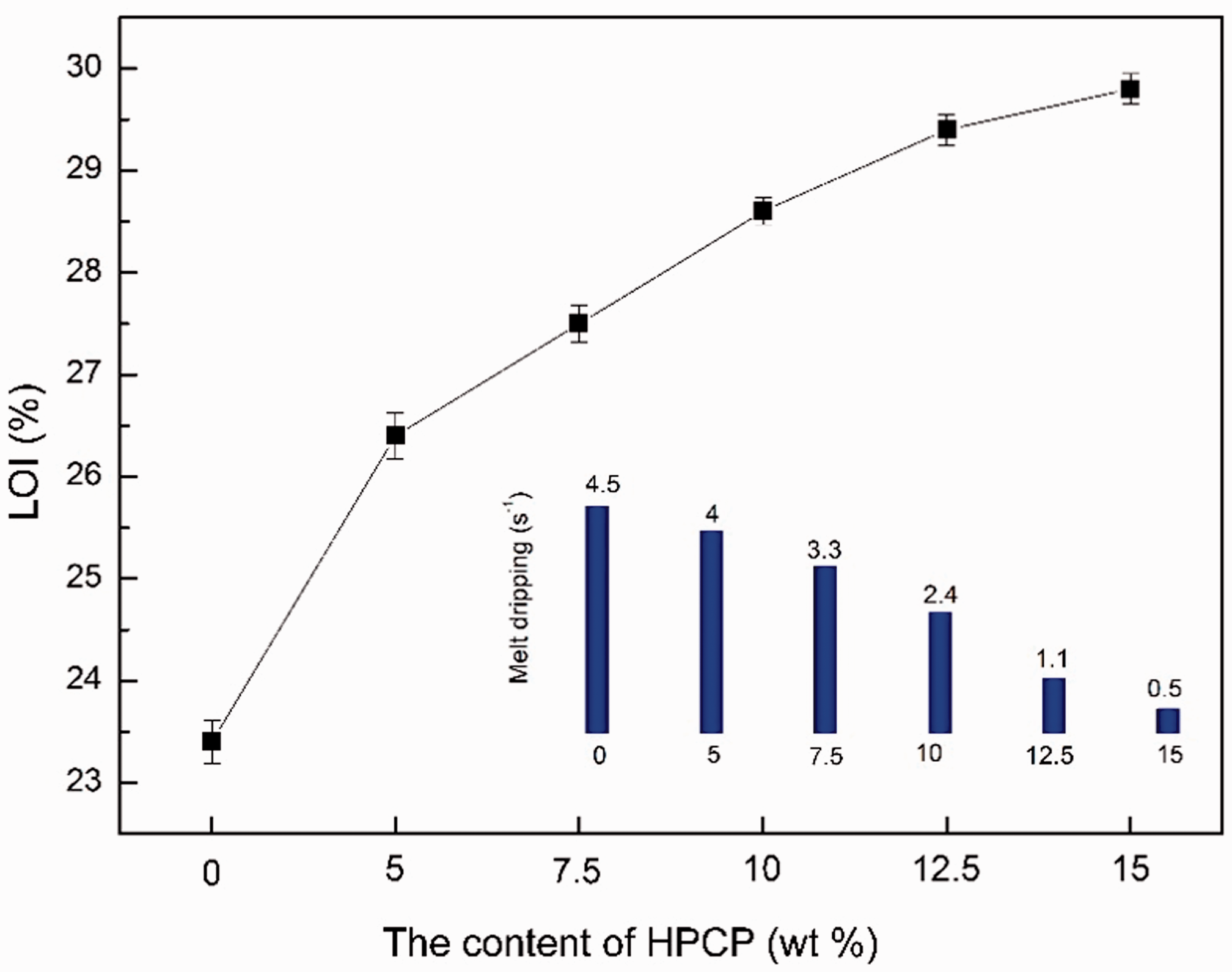
LOI value and melt dripping of the PA6 fiber samples.
To further evaluate the burning behavior of PA6/HPCP fibers, the effects of HPCP on afterflame time, afterglow time, damaged length and ignition times of the fibers tested by 45-degree slope burning are summarized in Table 5. Pure PA6 fibers ignited easily and combusted fast after the application of the flame accompanying many drips. The presence of HPCP significantly declined the burning time, indicating the lower content of flammable gases which are conductive to combust. The PA6/HPCP-5 and PA6/HPCP-7.5 can reach three ignitions which had the flame-retardant effect. With the 15 wt% content of HPCP, the fibers had no afterglow time and survived five ignitions which had excellent self-extinguished property. Based on the above results, HPCP has a positive flame-retarded effect on PA6 fibers.
Result of 45-degree slope burning test of the fibers.

Conclusions
The flame-retarded PA6 composite fibers were prepared successfully with HPCP by melt spinning. HPCP declined the melting point and melt viscosity of PA6 chips, which is beneficial to the spinning process. The spinning temperature and quench flow rate of PA6/HPCP fibers were lower than those of pure PA6 fibers. The achievable draw ratio at 4.6 had acceptable mechanical property for PA6/HPCP fibers. HPCP had no influence on the crystal configuration of the PA6 fibers in which the γ-crystal phase was predominant. With the addition of 10 wt% HPCP, the breaking tenacity of PA6 fibers decreased by 14%. The orientation factor values showed a moderate downtrend trend with the increase in HPCP content. The residue of PA6/HPCP fibers was increased. PA6 fibers reached an LOI value at 29.8 and survived five ignitions with no afterglow time when the HPCP content was 15 wt%. Therefore, PA6/HPCP fibers with comprehensive performances are promising materials for functional textile applications.
Footnotes
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This project was supported by the Natural Science Foundation of Shanxi, China (Grant No. 201801D121279) and Graduate Innovation Program of Shanxi China (Grant No. 2019SY107).