
Abstract
Aims:
Plasma-activated liquid (PAL), an indirect application form of cold-atmospheric plasma (CAP)—an ionized gas generating reactive oxygen and nitrogen species, has been proposed as an innovative therapeutic approach for various cancer types. Despite accumulating evidence suggesting that PAL induces cell death through multiple mechanisms, the involvement of ferroptosis, a form of cell death driven by iron and lipid peroxidation, in osteosarcoma (OS) remains predominantly unknown.
Results:
CAP was used to activate the liquid for various durations, resulting in different doses of PAL. The antitumor efficacy of PAL was directly correlated with both the dosage and duration of treatment and was achieved by increasing the level of intracellular reactive oxygen species. Through screening three effective PAL doses, we discovered that PAL significantly influenced the migration and invasion capabilities of OS cells. Proteomic sequencing revealed increases in several ferroptosis-related antioxidant proteins in the PAL-treated group. Subsequent findings revealed that PAL modulated nuclear factor erythroid 2-related factor 2 (NRF2) and its downstream ferroptosis-related genes, predominantly resulting in the induction of ferroptosis by depleting glutathione peroxidase 4 (GPX4) in human OS cells. Finally, utilizing an OS xenograft model, we found that PAL effectively suppressed tumor growth in vivo via ferroptosis.
Innovation:
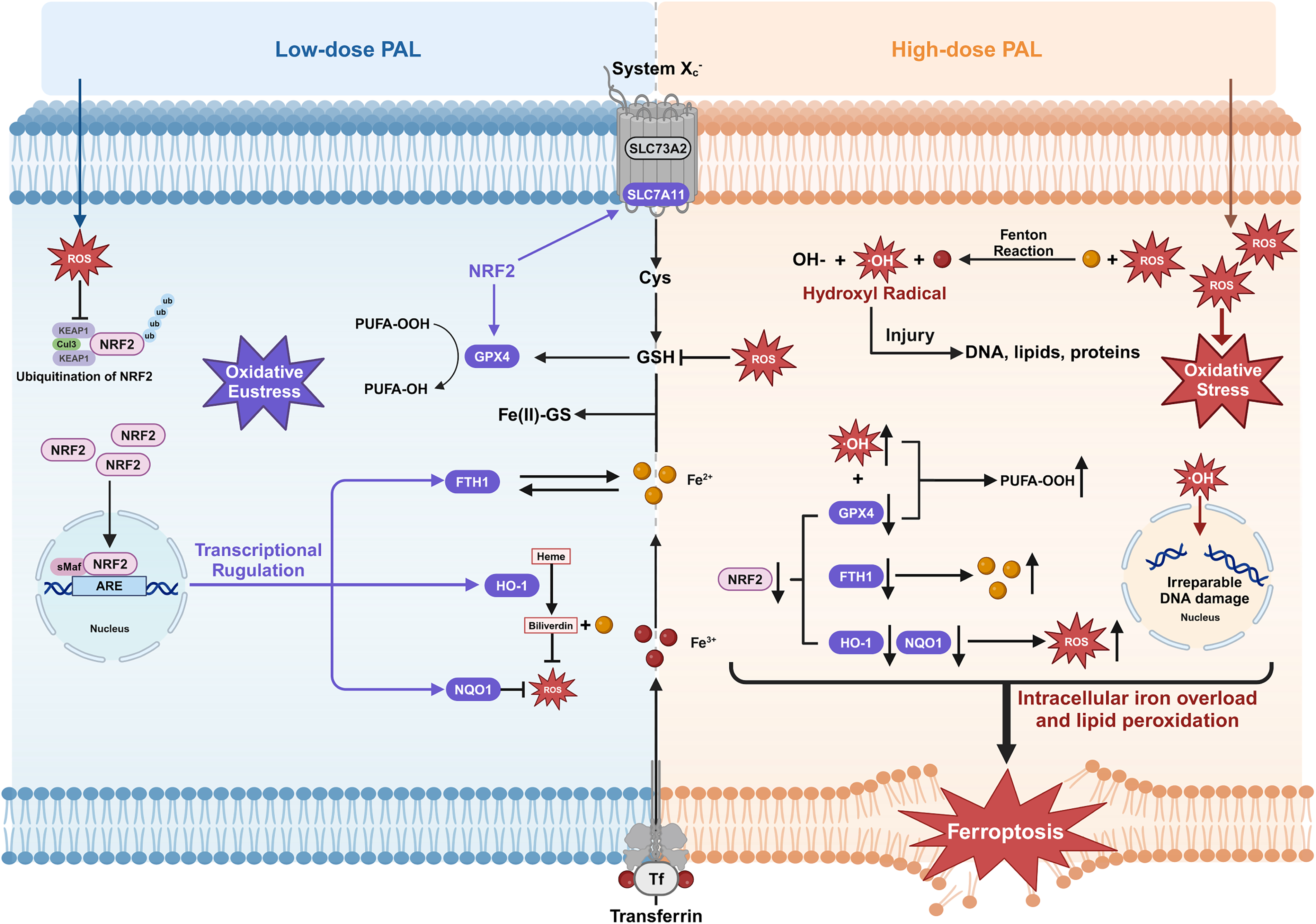
Our study highlights the importance of the NRF2/GPX4 axis as a pivotal pathway in PAL-induced ferroptosis. In vivo experiments provided compelling evidence supporting the potential of PAL as a potent therapeutic strategy for OS treatment.
Conclusion:
High-dose PAL-induced sustained oxidative stress by simultaneously targeting NRF2 inactivation and GPX4 degradation, establishing redox imbalance as a critical ferroptotic checkpoint in OS therapy. Antioxid. Redox Signal. 44, 292–310.
Keywords
Introduction
Osteosarcoma (OS), the most common primary malignant bone tumor, predominantly affects adolescents and young adults, with an annual incidence of 3–5 cases per million people (He et al., 2023; Ritter and Bielack, 2010). Its aggressive nature manifests through rapid local invasion and early pulmonary metastasis, contributing to a 5-year survival rate of less than 70% despite multimodal therapies. Current standard of care combines limb-salvage surgery with neoadjuvant polychemotherapy regimens (typically methotrexate, doxorubicin, and cisplatin) and radiotherapy (Gill and Gorlick, 2021). While this approach has improved the 5-year survival from <20% in the prechemotherapy era to 60%–70% today, nearly 30% of patients develop chemoresistance within 2 years, culminating in lethal relapse. Therefore, identifying a novel therapeutic approach capable of addressing the issue of chemoresistance in individuals with OS is imperative (Chen et al., 2021a; Suit, 1975).
Innovation
Plasma-activated liquid (PAL), which leverages stabilized reactive species, shows promise in treating solid tumors, yet its role in modulating ferroptosis and malignant progression remains underexplored. While previous studies have linked prolonged PAL exposure to FSP1-dependent ferroptosis resistance, we demonstrated that high-dose PAL suppressed osteosarcoma (OS) migration, invasion, and growth via dose-dependent NRF2/GPX4 axis disruption, inducing a redox imbalance and ferroptosis. The results of this study shift the paradigm from treatment duration to dosage optimization, revealing the dual-phase activity of PAL, namely, the low-dose adaptive antioxidant response versus high-dose ferroptotic cell death, offering a novel therapeutic strategy to overcome chemoresistance and metastasis in OS.
Emerging as a frontier in oncotherapy, cold-atmospheric plasma (CAP) is a state of ionized gas that closely approximates room temperature and encompasses a variety of reactive species, including ions, electrons, neutral particles, and visible UV photons (Fridman et al., 2008; Ishaq et al., 2014). The tumor selectivity of CAP stems from differential redox buffering capacities: malignant cells exhibit elevated basal reactive oxygen species (ROS) levels due to metabolic reprogramming, rendering them vulnerable to reactive oxygen/nitrogen species (RONS)-induced oxidative stress. Mechanistically, CAP-derived RONS (e.g., •OH, O2-, NO•, and H2O2) disrupt redox homeostasis through mitochondrial membrane depolarization, DNA strand breaks, and lipid peroxidation cascades (Dai et al., 2023; Fang et al., 2023; Graves, 2017). However, direct CAP application faces critical limitations: its reactive species exhibit ultrashort half-lives (nanoseconds to milliseconds) and limited tissue penetration depth (<2 mm), restricting its utility to superficial malignancies such as cutaneous squamous cell carcinoma or oral premalignant lesions (Iuchi et al., 2023; Yan et al., 2015).
This technological barrier has spurred the development of plasma-activated liquid (PAL)-CAP-treated aqueous solutions that stabilize CAP-derived RONS into long-lived derivatives (e.g., NO2-, NO3-, O3) through complex aqueous-phase reactions (Tanaka et al., 2016; Tornín et al., 2023; Xu et al., 2023.). While CAP has gained traction in dermatology and wound healing due to its superficial applications, its indirect form, PAL, offers a novel approach for oncology by stabilizing RONS through plasma–liquid interactions (Bruggeman et al., 2016). Unlike radiolyzed or ozonated water (Tanaka et al., 2016), which may produce similar species such as H2O2 or O3, PAL exhibits a distinct RONS profile (e.g., elevated nitrite, nitrate, and peroxynitrite) that arises from complex aqueous-phase reactions, potentially enhancing selectivity for cancer cells through mechanisms such as ferroptosis. Although large-scale clinical trials in oncology are still pending, preliminary studies support PAL’s potential in overcoming chemoresistance, particularly in aggressive cancers such as OS. PAL retains the tumoricidal properties of CAP while overcoming penetration constraints, enabling deep-seated tumor targeting. Notably, our preliminary work revealed the unique dual-phase activity of PAL in OS: Low-dose PAL initially activated the antioxidant response in cells until the cells died due to oxidative imbalance, whereas high-dose administration triggered catastrophic oxidative stress.
Ferroptosis is characterized by iron-dependent lipid peroxidation, ultimately resulting in cellular death (Chen et al., 2021b; Dixon et al., 2012). Seminal work by Yang et al. and Friedmann Angeli et al. identified glutathione peroxidase 4 (GPX4) as the central regulator of ferroptosis, demonstrating that its inhibition is sufficient to trigger this form of cell death (Friedmann Angeli et al., 2014; Yang et al., 2014). Nuclear factor erythroid 2-related factor 2 (NRF2) rectifies the cellular redox imbalance through the regulation of iron, glutathione (GSH), and ROS levels via the modulation of numerous downstream target genes (Dodson et al., 2019). Previous studies have identified GPX4 and ferroptosis suppressor protein 1 (FSP1) as pivotal regulators that exert inhibitory effects on ferroptosis. GPX4 and FSP1 are involved in conferring resistance to ferroptosis and lipid peroxidation through parallel and independent systems: GPX4 utilizes GSH to eliminate lipid peroxides generated in phospholipids containing polyunsaturated fatty acids (PUFAs), while FSP1 localizes to the plasma membrane, where it functions as an oxidoreductase that reduces coenzyme Q10, generating a lipophilic radical-trapping antioxidant that halts the propagation of lipid peroxides (Bersuker et al., 2019; Doll et al., 2019; Stockwell, 2022).
The rich repertoire of RONS in PAL initiates iron-mediated Fenton chemistry within tumor cells, effectively overwhelming endogenous antioxidant defenses (Suzuki-Karasaki et al., 2023). This redox perturbation triggers a self-amplifying cascade characterized by mitochondrial depolarization, dysregulated iron homeostasis, and lethal lipid peroxide accrual, which are hallmarks of ferroptotic death (Stockwell et al., 2017; Toyokuni et al., 2017). By using a CAP activation time-dependent dose–escalation design, we generated PAL formulations with graduated bioactivity. Proteomic profiling revealed the paradoxical upregulation of NRF2-regulated heme oxygenase-1 (HO-1) under low-dose PAL exposure, suggesting compensatory antioxidant adaptation (Dodson et al., 2019; He et al., 2020). Systematic assessments of ferroptosis-related effectors across dosage gradients demonstrated biphasic regulation: High-dose PAL suppressed NRF2 nuclear translocation while promoting GPX4 proteasomal degradation, effectively dismantling the GSH-dependent antioxidant axis. Crucially, subcutaneous xenograft models confirmed in vivo tumor suppression through GPX4 downregulation-mediated ferroptosis. Our findings definitively demonstrate that high-intensity PAL therapy subverts the NRF2/GPX4 signaling nexus to establish a proferroptotic tumor microenvironment (Graphical abstract).
Results
PAL-mediated cytotoxic efficacy in OS cells is dose and time dependent
To investigate the potential of PAL to induce cell death in human OS cells and identify factors influencing its antitumor efficacy, we exposed complete medium to CAP (Fig. 1A) for various durations to generate different doses of PAL. Subsequently, SAOS-2 and U2OS cells were treated with various doses of PAL for 12 or 24 h. Our results demonstrated that the cytotoxic effect of PAL on both the OS cell lines was dose dependent (Fig. 1B). In addition, we observed a positive correlation between the duration of PAL treatment and its cytotoxic effect on tumor cells, with cell viability being significantly reduced after 24 h of continuous exposure.
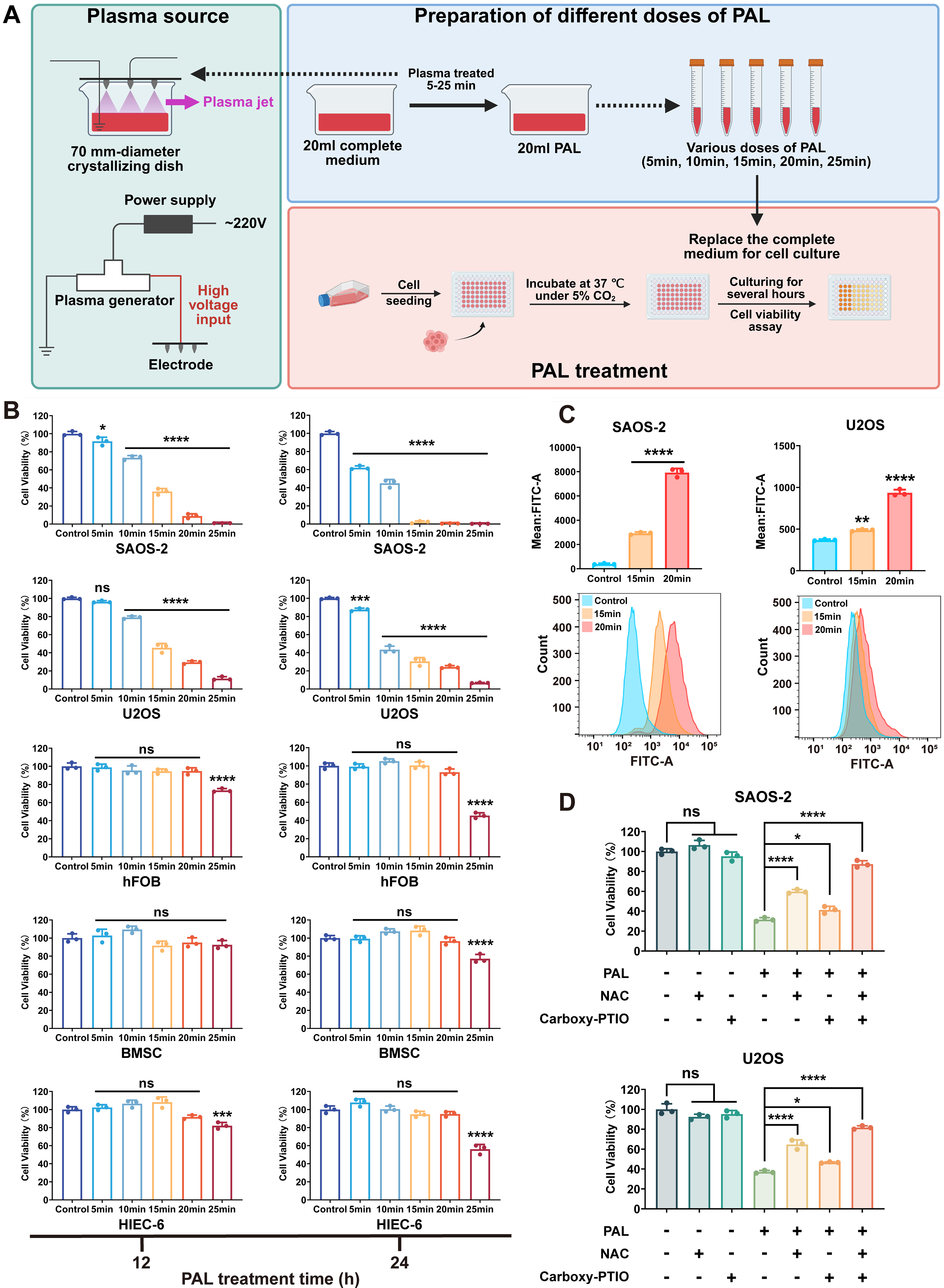
To identify effective PAL doses that kill OS cells without affecting normal cells, experiments revealed that treatment with PAL5min for 12 h had no significant effect on U2OS cells. Moreover, PAL10min, PAL15min, and PAL20min did not affect the activity of human osteoblast (hFOB), human bone marrow mesenchymal stem cells (BMSCs), and human intestinal epithelial cells (HIEC-6), whereas PAL25min had a significant inhibitory effect on these normal cells (Fig. 1B). The effective doses of PAL were 10, 15, and 20 min.
PAL-induced cancer cell death is correlated with elevated levels of ROS
PAL’s complex RONS composition may simultaneously activate other programmed death pathways, including apoptosis, necrosis, autophagy, ferroptosis, and pyroptosis (Azzariti et al., 2019; Moloney and Cotter, 2018), depending on treatment parameters and cellular redox status. The dominant death mechanism is determined by an integrative effect of treatment intensity, duration, and target cell vulnerability. Critically, longitudinal ROS assessment via fluorescence microscopy (Supplementary Fig. S2A and B) revealed progressive accumulation patterns in both cell lines: ROS levels escalated with extended PAL exposure (4→8→12 h), exhibiting strict both concentration and temporal dependence. This oxidative surge reached a critical threshold in the PAL20min treatment by 8–12 h, coinciding precisely with irreversible viability loss, where cell survival decreased to below 40% (Fig. 1B). At this point, the oxidative burden definitively overwhelmed cellular adaptive capacity, triggering the collapse of redox homeostasis.
To further elucidate the contribution of reactive species, we evaluated the effects of scavengers: N-acetylcysteine (NAC, an ROS scavenger) and carboxy-PTIO (an RNS scavenger). Both scavengers significantly reversed the PAL-induced decrease in tumor cell viability, but NAC exhibited a stronger protective effect compared with carboxy-PTIO (Fig. 1D). This indicates that while both ROS and RNS are involved in PAL-induced cytotoxicity, ROS play a more dominant role in driving cell death.
The PAL dosage gradient regulates the long-term proliferation of OS cells
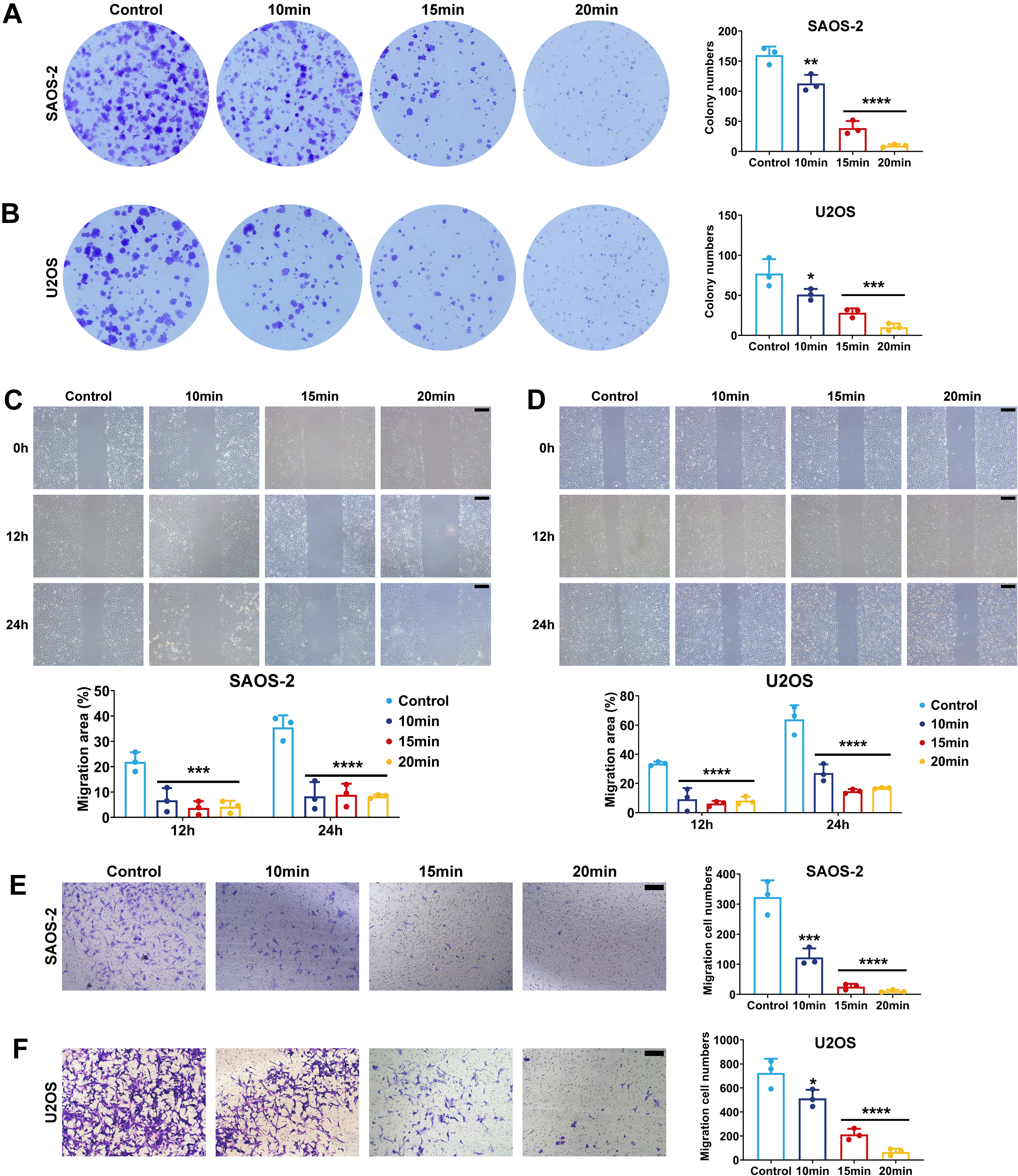
Based on prior cell viability assays, we identified CAP treatment durations of 10, 15, and 20 min as critical thresholds. These doses significantly inhibited the short-term proliferation of OS cells while preserving normal cell viability. To evaluate the impact of PAL on long-term proliferation and clonogenicity, two OS cell lines were pretreated with PAL10min, PAL15min, or PAL20min for 12 h. Equal numbers of untreated and pretreated cells were then seeded in six-well plates. After 2 weeks, the colony formation assay results combined with the CCK-8 assay results demonstrated a significant positive correlation between the PAL dose gradient and the suppression of tumor proliferation (Fig. 2A and B).
PAL suppresses the malignant progression of OS
Pulmonary metastasis is a critical determinant of poor OS. To model early steps in metastasis, we performed scratch assays to simulate local tissue migration and Transwell invasion assays to mimic basement membrane penetration—key processes in metastatic dissemination. In this study, we systematically evaluated the regulatory effects of effective PAL (PAL10min, PAL15min, and PAL20min) on OS cell migration and invasion through scratch assays and Transwell invasion assays. Scratch assays demonstrated that compared with the control, PAL pretreatment (12 h) significantly reduced the migration rates of OS cells at both the 12 and 24-h time points (Fig. 2C and D), with inhibition occurring in a dose-dependent manner. Although the observed reduction in scratch assays could be partially influenced by decreased cell viability due to PAL treatment, the consistent dose-dependent suppression of migration and the complementary results from Transwell invasion assays (which are less affected by viability changes) suggest that PAL directly impairs the migratory capacity of OS cells independently of cytotoxicity. Transwell invasion assays confirmed that PAL treatment markedly suppressed the invasive capacity of OS cells (Fig. 2E and F). PAL pretreatment significantly inhibited both migration and invasion in OS cells (Fig. 2C–F), suggesting its potential to suppress the initial stages of metastasis.
PAL-induced cell death involves multiple antioxidant pathways linked to ferroptosis
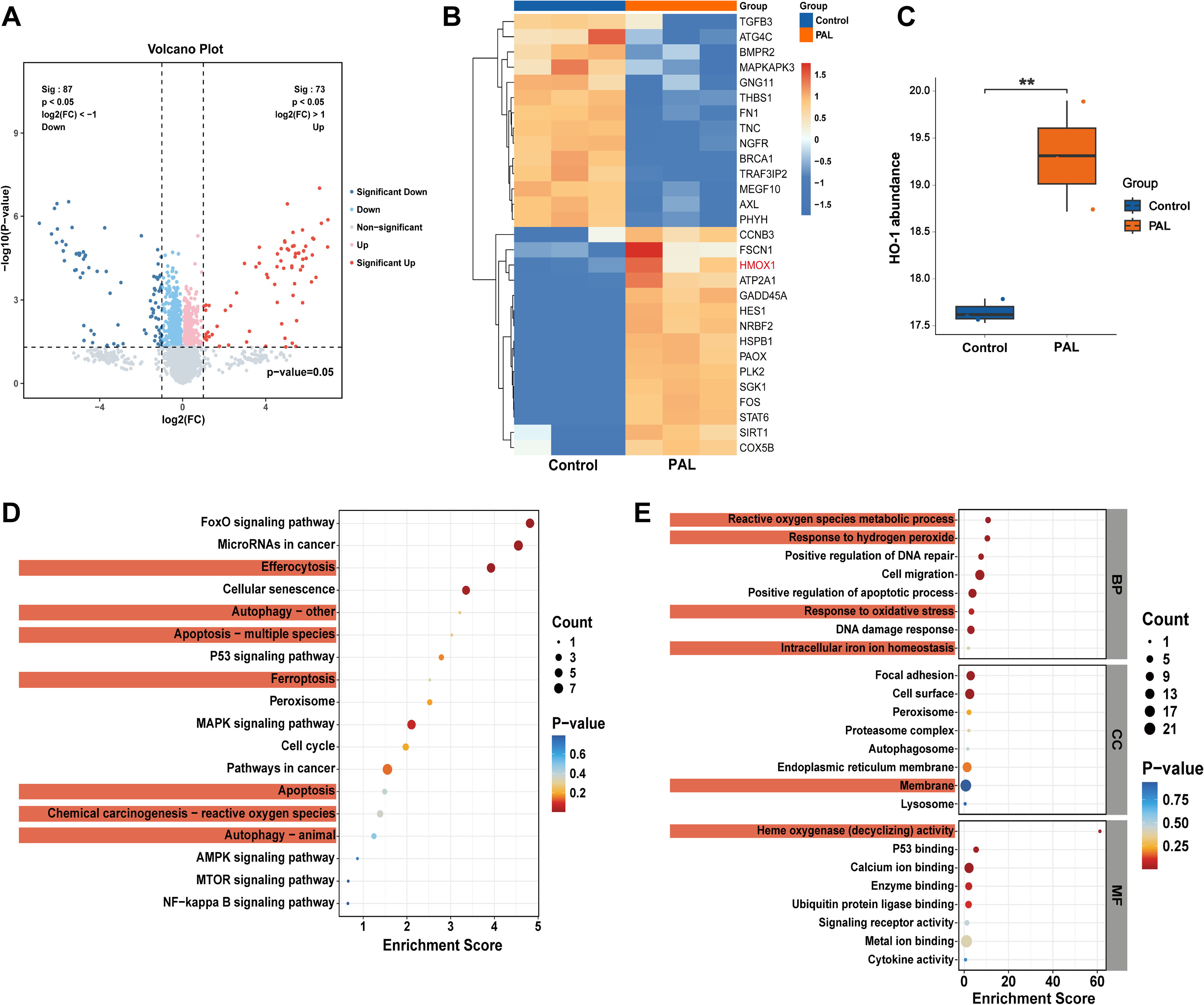
To investigate the mechanism of PAL-induced cell death, we performed proteomic analysis on both SAOS-2 cells treated with PAL10min for 12 h and untreated SAOS-2 cells. The findings revealed significant alterations in genes associated with oxidative stress in the low-dose PAL group (Fig. 3A, D, and E), particularly a marked upregulation of the HO-1 gene (Fig. 3B and C). Notably, HO-1 serves as a downstream effector of the antioxidant transcription factor NRF2, facilitating the breakdown of heme into bilirubin and Fe2+. As a regulator of intracellular free iron and ROS levels, HO-1 plays a crucial role in modulating ferroptosis (Sun et al., 2016).
Different doses of PAL-induced alterations in the KEAP1/NRF2 and SLC7A11/GPX4 signaling pathways in OS cells
Previous proteomic results demonstrated that PAL10min upregulated the expression of ferroptosis-related antioxidant genes in SAOS-2 cells. To elucidate the mechanism by which PAL mediates ferroptosis, we examined the expression levels of ferroptosis-related marker proteins in two OS cell lines using Western blot analysis (Fig. 4A).
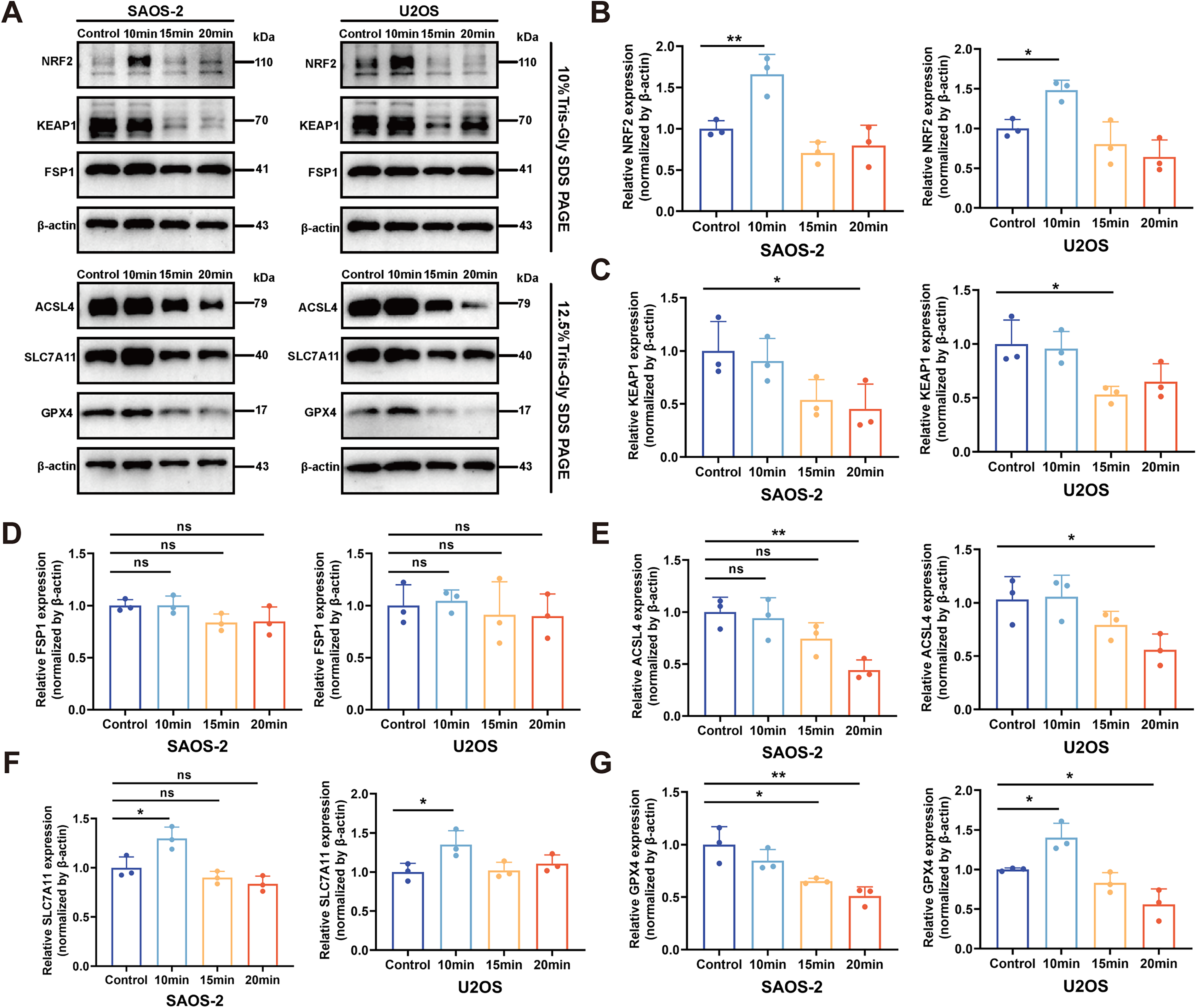
As a regulator of NRF2, KEAP1 typically induces the ubiquitination and degradation of NRF2, resulting in low basal protein levels of NRF2 under nonoxidative stress conditions (Sun et al., 2016). Upon exposure to oxidative stress, ROS trigger modifications of cysteine residues within KEAP1 and suppress its expression, allowing NRF2 to evade Keap1-mediated degradation (Ross and Siegel, 2021). As a result, newly synthesized NRF2 accumulates in the nucleus where it regulates downstream target gene expression and maintains redox balance (Dodson et al., 2019; Sies, 2021). The observed gradual decrease in KEAP1 expression with increasing doses of PAL in OS cell lines suggests a dose-dependent exacerbation of oxidative stress (Fig. 4C). The dissociation between KEAP1 reduction and NRF2 induction at high PAL doses reflects the overarching impact of oxidative overload, which uncouples canonical regulation and leads to pathway failure.
NRF2 and SLC7A11 protein expression was upregulated in both OS cell lines following low-dose PAL treatment (Fig. 4B and F). Conversely, high-dose PAL treatment significantly reduced the levels of NRF2 and GPX4 in OS cells (Fig. 4B and G). The SLC7A11/GPX4 axis plays a crucial role in the NRF2-mediated regulation of GSH metabolism. These findings indicate that low-dose PAL-induced oxidative stress appropriately stimulates NRF2, leading to increased SLC7A11 expression and protection against ferroptosis. In contrast, a high dose of PAL disrupts NRF2 levels, ultimately inhibiting GPX4 expression. Subsequent analysis demonstrated that treatment with PAL under identical conditions did not significantly alter the expression of the NRF2/GPX4 pathway in hFOB, BMSC, or HIEC-6 normal cell lines (Supplementary Fig. S3), which—combined with the absence of cytotoxicity observed in cell viability assays (Fig. 1B)—collectively confirms the minimal toxicity of the selected PAL dosage toward nonmalignant cells.
The acyl-CoA synthetase long chain family member 4 (ACSL4) protein is a key regulator that promotes ferroptosis by enriching membranes with peroxidation-susceptible PUFAs (Stockwell et al., 2017). Although high-dose PAL treatment reduced ACSL4 expression (Fig. 4E)—a change potentially reflecting compensatory stress adaptation—this effect was functionally overridden by dominant GPX4 suppression. Critically, >50% GPX4 depletion (Fig. 4G) disrupted GSH-dependent lipid peroxide clearance, overwhelming any antiferroptotic influence from ACSL4 downregulation. This aligns with established models where GPX4 ablation suffices to induce ferroptosis irrespective of ACSL4 status.
Furthermore, the antiferroptosis pathways mediated by FSP1 and GPX4 operate through mechanistically distinct, parallel systems. Notably, FSP1 expression remained unchanged across all PAL doses (Fig. 4D), indicating that PAL specifically targets the NRF2/GPX4 axis without engaging the FSP1/CoQ10 pathway. This selectivity is clinically advantageous, as it minimizes opportunities for ferroptosis escape mechanisms often observed with single-pathway inhibitors.
High-dose PAL depletes the expression of NRF2 and downstream antioxidant genes
NRF2, a stress-responsive transcription factor, translocates to the nucleus and activates the transcription of genes containing antioxidant response elements when degradation is hindered due to endogenous stress, genetic mutations, or exogenous drug inhibition (He et al., 2020). This regulatory mechanism plays a pivotal role in safeguarding cells against ferroptosis by orchestrating the upregulation of the expression of multiple genes, including HO-1, ferritin heavy chain 1 (FTH1), and quinone oxidoreductase-1 (NQO1) (Ross and Siegel, 2021), involved in iron and ROS metabolism.
In this study, we investigated whether increased NRF2 expression promotes the translocation of NRF2 to the nucleus and upregulates the expression of downstream target genes. Confocal laser scanning microscopy revealed a significant increase in NRF2 fluorescence intensity within the nucleus following low-dose PAL treatment (Fig. 5A and B). Consistent with previous proteomic findings, we observed upregulated HO-1 expression after low-dose PAL treatment (Fig. 5C and D). After low-dose PAL treatment, the expression of HO-1, NQO1, and FTH1 increased, whereas high-dose PAL stimulation led to a decrease in the expression of these three proteins, a result that was consistent with the previously observed trend in NRF2 expression (Fig. 5B and D–F). The reduction in FTH1 expression indicates diminished intracellular iron storage and elevated levels of free iron, as evidenced by the observed increase in the intracellular Fe2+ in OS cells (Fig. 5G and H). These findings suggest that the expression level of NRF2 is positively correlated with its antiferroptotic transcriptional effect mediated by NRF2.
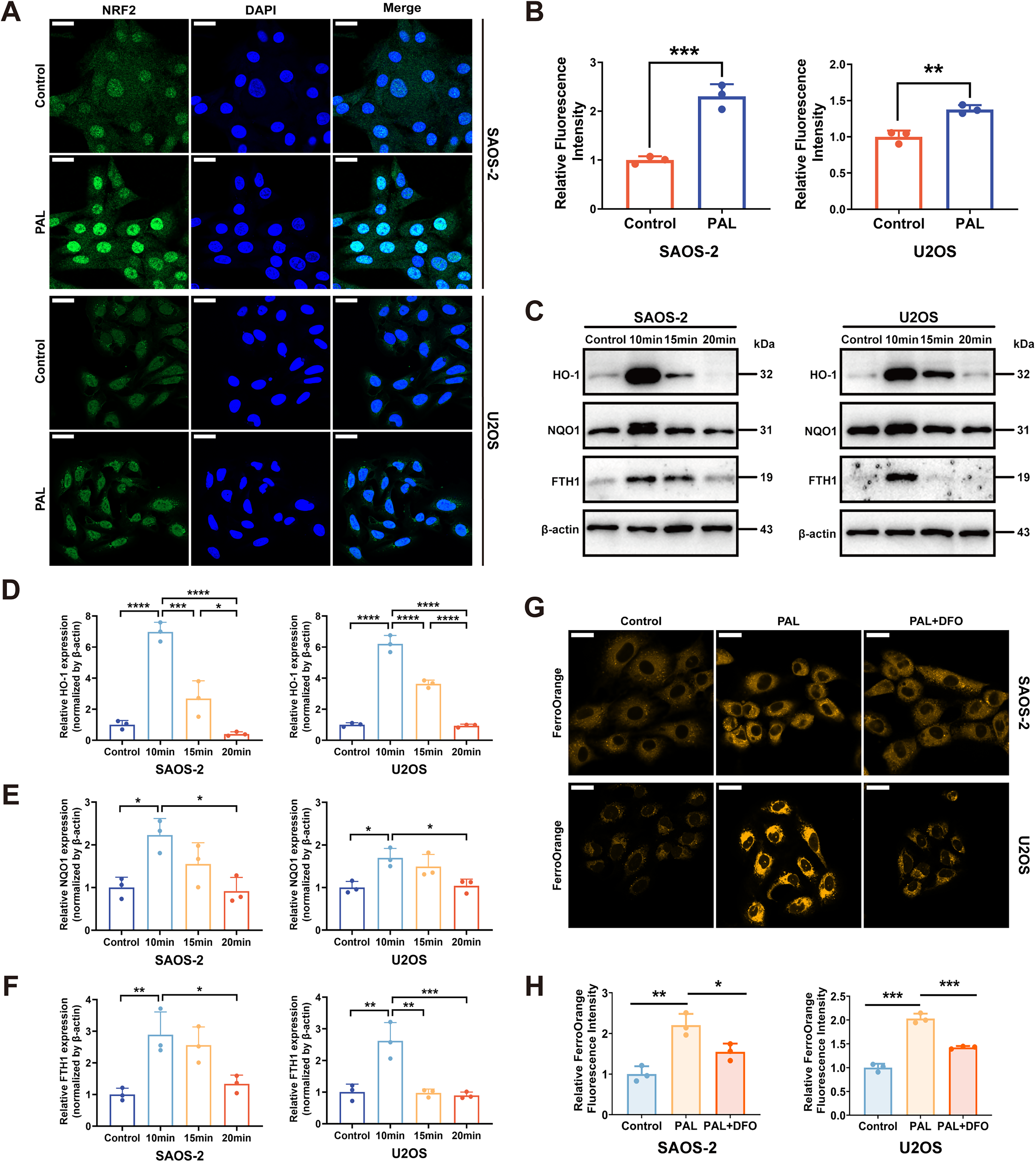
Overall, the expression of NRF2 and its downstream antioxidant genes was significantly downregulated following high-dose PAL treatment. These findings indicate that the oxidative stress induced by high-dose PAL impairs the capacity of NRF2 to maintain redox homeostasis, consequently leading to the reduced expression of downstream antioxidant genes.
High-dose PAL induces ferroptosis in OS cells by modulating the NRF2/GPX4 axis
Recent studies have identified ferroptosis as a form of cell death linked to oxidative stress that is predominantly driven by lipid peroxidation due to iron overload (Stockwell et al., 2017). Ferritin plays a crucial role in converting intracellular labile iron into its trivalent stored form (Hou et al., 2016). Our findings revealed that high-dose PAL downregulates GPX4 and FTH1 expression, thereby depleting iron reserves and consequently increasing the levels of labile iron (Fig. 5G and H).
Consequently, we hypothesized that high-dose PAL induces ferroptosis in OS cells. To validate this hypothesis, we examined additional hallmarks of ferroptosis. Specifically, the administration of the ferroptosis inhibitor ferrostatin-1 (Fe-1) or the iron chelator deferoxamine (DFO) both effectively reversed the PAL-induced decrease in OS cell viability (Fig. 6A and B). This increased cell viability was accompanied by a decrease in lipid peroxide levels, as evidenced by changes in the C11-BODIPY fluorescent probe intensity and malondialdehyde (MDA) content (Fig. 6C–E). Collectively, these findings provide compelling evidence that high-dose PAL induces ferroptosis in OS cells.
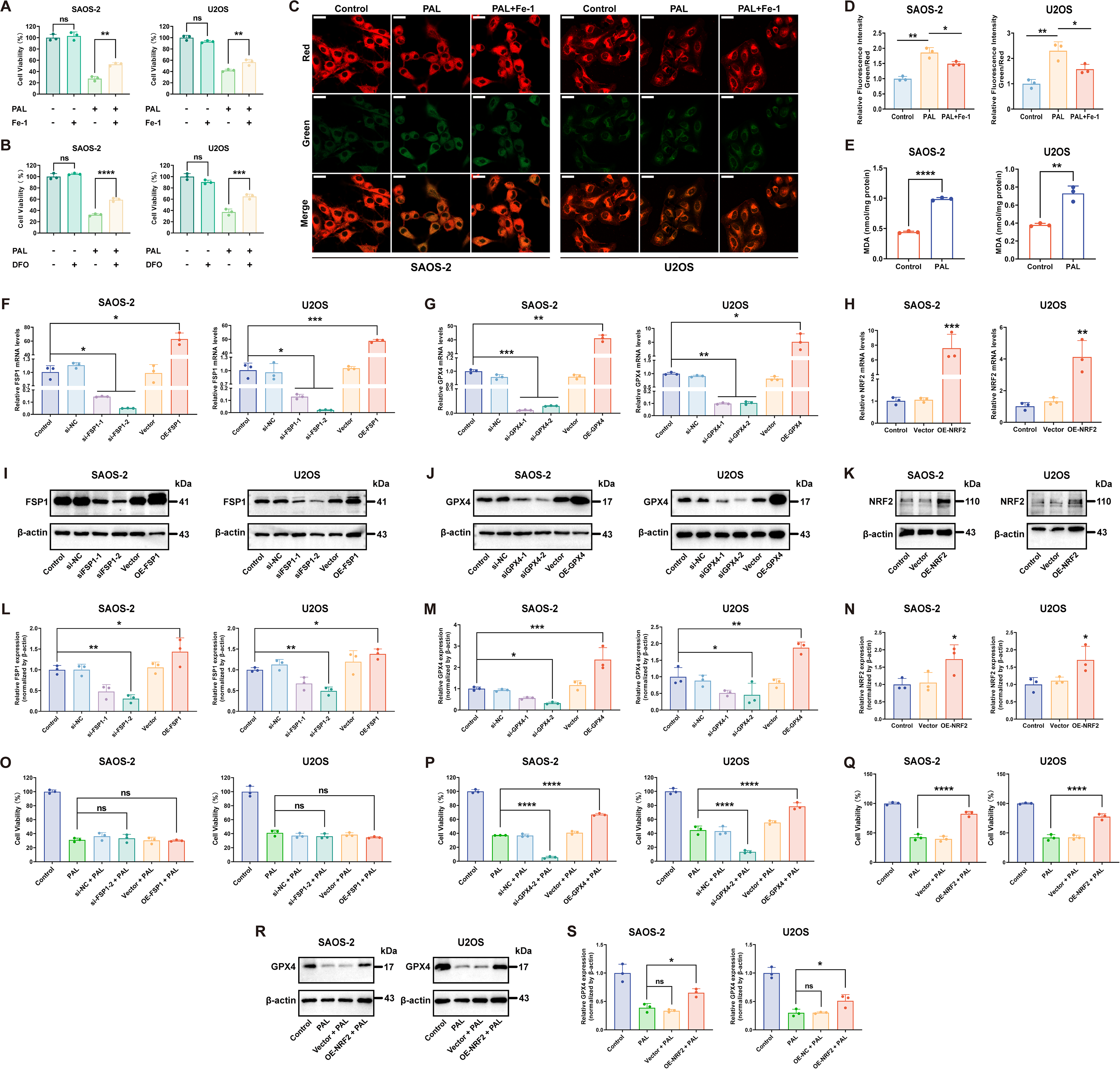
To verify the pathway specificity of PAL-induced ferroptosis, we genetically modulated FSP1 and NRF2/GPX4 axis expression in OS cells (Fig. 6F–N). Altering FSP1 expression had no effect on PAL sensitivity (Fig. 6O), whereas modulating GPX4 directly correlated with resistance (Fig. 6M). Moreover, NRF2 overexpression also increased PAL tolerance (Fig. 6Q), and these cells exhibited higher GPX4 levels after PAL challenge compared with untransfected cells (Fig. 6R and S). These findings conclusively show that high-dose PAL triggers ferroptosis primarily via the NRF2/GPX4 axis.
Time-resolved analysis of the NRF2/GPX4 axis revealed distinct adaptive patterns preceding ferroptotic commitment (Supplementary Fig. S4). In SAOS-2 cells, NRF2 increased at 4 h after high-dose PAL (PAL20min), followed by GPX4 reduction at 8 h under mid-to-high-dose exposure (PAL15min and PAL20min). U2OS cells exhibited delayed compensation: NRF2 elevation emerged only at 8 h with the PAL20min treatment, whereas GPX4 remained stable until 12 h. Critically, both lines converged at the 12-h threshold under the PAL20min upregulation with low-dose PAL (PAL10min) shifted to coordinated suppression of NRF2 and GPX4, directly correlating with the viability collapse point (survival <40%, Fig. 1B). This transition demonstrates that sustained high-dose PAL overrides compensatory mechanisms through irreversible NRF2/GPX4 axis failure.
Ferroptosis is involved in high-dose PAL-induced tumor suppression in vivo
To investigate whether high-dose PAL could inhibit tumor growth in vivo by inducing ferroptosis, we established a subcutaneous OS xenograft model in nude mice using SAOS-2 cells. Considering the potential impact of serum and antibiotics in complete medium on immunodeficient nude mice (Azzariti et al., 2019), we selected Ringer’s lactate (RL) solution as the liquid medium activated by CAP to formulate high-dose PAL for in vivo OS treatment (Jiang et al., 2021; Tanaka et al., 2016). Twenty-four 4-week-old male nude mice were randomly allocated into four groups: control group, negative control (NC) group, PAL group, and PAL pretreatment (Pp) group. Each mouse in the Pp group received a 100 μL injection of the RL mixture containing 5 × 106 PAL20min-pretreated SAOS-2 cells, which had been pretreated for 12 h, in the left axilla. The remaining three groups received a 100 μL injection of the RL mixture containing 5 × 106 untreated SAOS-2 cells at the same location. When the tumor size in each mouse from the control, NC, and PAL groups, which had been inoculated with untreated SAOS-2 cells, reached 100 mm³, the intervention treatment was initiated. Nude mice in the NC and PAL groups received intratumoral injections of 100 μL of RL or 100 μL of PAL20min. Subcutaneous tumor volume was measured and recorded before each intratumoral injection. Nude mice in the control and Pp groups did not undergo any intervention during the treatment period (Fig. 7A).
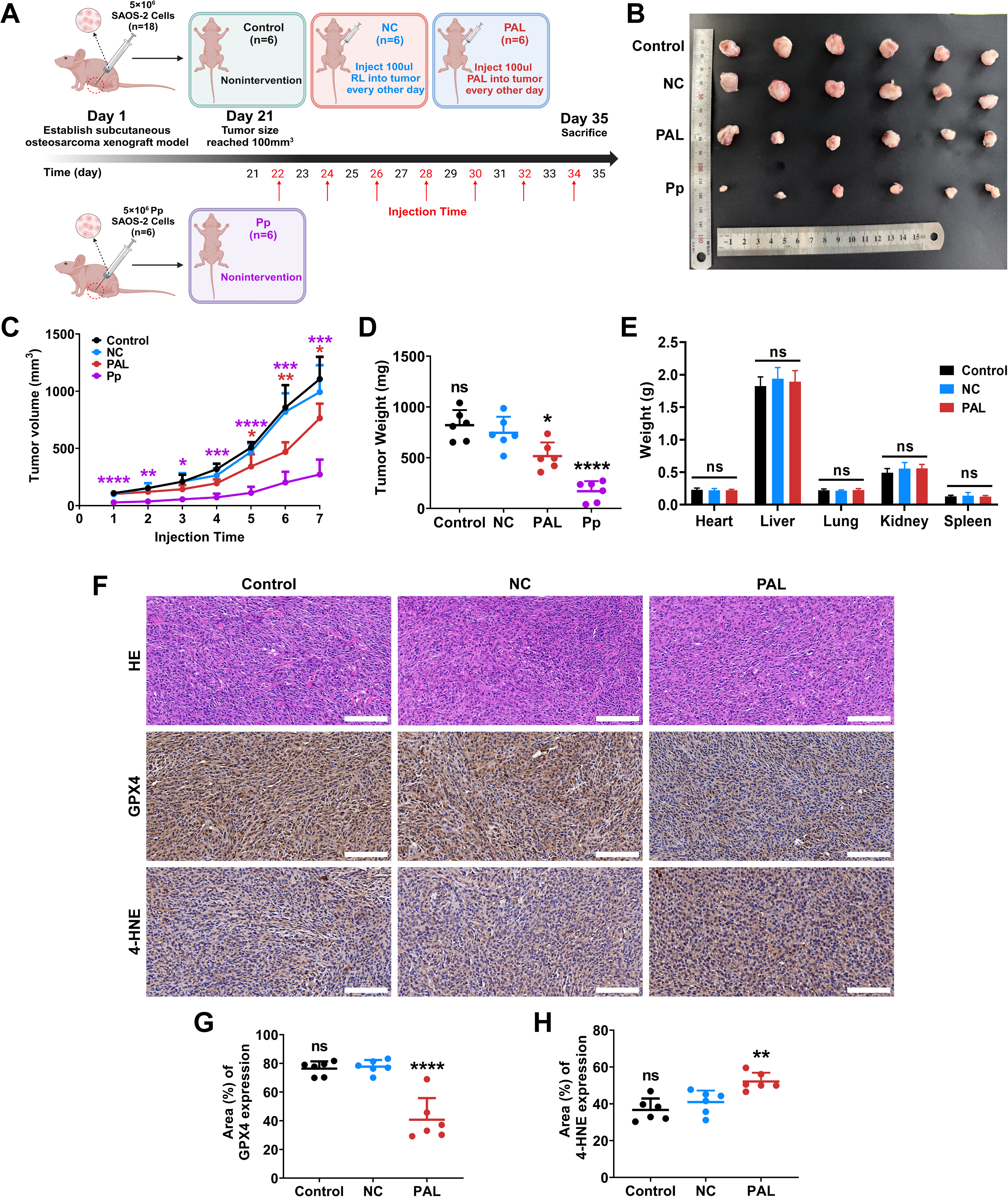
The results indicated that there were no significant differences in tumor volume or weight between the control group and the NC group that received RL injection, demonstrating that the intratumoral injection of RL did not impact tumor growth. Compared with that in the NC group, the tumor size in the PAL group significantly differed, with a final tumor weight that was significantly lower than that in the NC group. Moreover, both the growth rate and final tumor volume in the Pp group differed significantly from those in the NC group (Fig. 7B–D), indicating that cells pretreated with PAL exhibited markedly restricted malignant progression over an extended period, even in the absence of subsequent PAL treatment. The Pp group was included to evaluate the duration of PAL-induced effects ex vivo, demonstrating that even transient pretreatment can substantially impair tumorigenicity, supporting potential applications in surgical adjuvant settings.
These findings suggest that PAL profoundly influences the OS growth rate and size in vivo. Finally, assessments of hematoxylin–eosin (HE) staining and vital organ weights also demonstrated the absence of any adverse effects on the nude mice following treatment (Fig. 7E and Supplementary Fig. S6). The immunohistochemistry results demonstrated that PAL decreased GPX4 staining intensity in tumor samples while increasing the staining intensity of 4-hydroxynonenal (4-HNE) (Fig. 7F–H), the end product of lipid peroxidation. In the subsequent OS xenograft models, animals were randomized into PAL and PAL + Fe-1 groups (ferroptosis inhibitor Fe-1), following the experimental scheme outlined in Figure 7A. Results demonstrated that after the fourth administration, tumor volumes were significantly reduced in the PAL group compared with the PAL + Fe-1 group (Supplementary Fig. S5B). Endpoint analyses confirmed markedly smaller tumor size and lower weight in the PAL group (Supplementary Fig. S5A and C). Immunohistochemical staining revealed that Fe-1 effectively reversed PAL-induced suppression of GPX4 and elevation of 4-HNE (Supplementary Fig. S5D and E), underscoring that ferroptosis inhibition abrogates the antitumor effects of PAL via the GPX4 pathway. Importantly, while PAL may potentially trigger multiple cell death modalities under different experimental conditions, these in vivo findings emphasize that ferroptosis through NRF2/GPX4 axis disruption remains the dominant and specific mechanism in our current setup, as evidenced by the comprehensive rescue effects of Fer-1. Collectively, our findings indicate that high-dose PAL can restrict tumor growth through ferroptosis.
Discussion
CAP, an ionized physical agent that generates diverse reactive species (Bauer et al., 2019; Dai et al., 2022), exhibits selective cytotoxicity toward malignant cells while sparing normal tissues, positioning it as a promising strategy to circumvent therapeutic resistance in oncology (Bruggeman et al., 2016; Yan et al., 2015). Although CAP has been shown to induce multiple programmed cell death modalities (Wang et al., 2022), the mechanisms underlying its PAL-mediated effects remain underexplored. Emerging evidence suggests that CAP-activated liquids—including cell culture medium (Adachi et al., 2016; Jo et al., 2022), phosphate-buffered saline, and lactated Ringer’s solution (Jiang et al., 2021)—retain antitumor efficacy. Our study used complete medium for in vitro PAL experiments to maintain cellular homeostasis while opting for serum-free RL solution in xenograft models to avoid immune interference. Both formulations demonstrated potent tumor-suppressive effects in their respective experimental systems.
Notably, PAL-induced cytotoxicity exhibited dose- and time-dependent correlations with intracellular ROS accumulation, which aligns with prior reports on reactive species dynamics during PAL exposure (Bersuker et al., 2019; Conway et al., 2016; Yan et al., 2015). Our findings indicate that the partial reversal of PAL-induced cell death by NAC underscores the crucial but not exclusive role of ROS in this process. Further investigation reveals that RNS also contribute to PAL’s cytotoxicity, as demonstrated by the synergistic protective effect observed when combining NAC with the RNS scavenger carboxy-PTIO. This synergy nearly abolishes PAL-induced cell death, highlighting that both ROS and RNS are integral components of PAL’s reactive species repertoire, working in concert to overwhelm cellular defenses and trigger ferroptosis. Thus, PAL’s antitumor effects are mediated by a complex interplay of RONS, which may involve cross talk between the oxidative and nitrosative stress pathways, providing a more comprehensive mechanistic understanding beyond ROS alone. To dissect dose-specific mechanisms, proteomic profiling of low-dose PAL-treated cells revealed the marked upregulation of the expression of HO-1, a canonical NRF2-regulated antioxidant gene. Subsequent confocal imaging confirmed enhanced nuclear NRF2 translocation and the coordinated induction of downstream targets (HO-1, FTH1, and NQO1) under low-dose conditions, suggesting adaptive antioxidant activation. Paradoxically, high-dose PAL suppressed these protective responses, indicating NRF2 pathway exhaustion or inactivation.
Our findings demonstrate that PAL induces ferroptosis in OS via NRF2/GPX4 axis disruption. Importantly, this study also reveals that low-dose PAL elicits a hormetic response, characterized by the activation of NRF2-mediated antioxidant pathways and upregulation of vitagenes (e.g., HO-1). This adaptive mechanism enhances cellular resilience against oxidative stress and may have broader implications for neuroprotection, as vitagene networks are known to mitigate protein misfolding and mitochondrial dysfunction in neurodegenerative contexts (Calabrese et al., 2010). Future studies should explore PAL’s potential as a hormetic agent in neurological models.
Given the reliance of ferroptosis on iron-dependent oxidative cascades, the observed FTH1 downregulation and redox imbalance following high-dose PAL treatment suggest this death modality. Biochemical analyses confirmed hallmark features: elevated labile iron pools and lipid peroxides. Intriguingly, NRF2 exhibited a biphasic expression pattern—initial upregulation followed by progressive suppression with increasing PAL doses—which directly influenced the SLC7A11/GPX4 antioxidant axis. Importantly, GPX4 depletion emerged as the linchpin mediating PAL-triggered ferroptosis, an effect that was corroborated by both in vitro sensitivity assays and in vivo tumor suppression via GPX4 downregulation.
Diverging from Ara Jo’s observation of FSP1-dependent ferroptosis without GPX4 modulation in lung cancer models under prolonged PAL exposure (Jo et al., 2022), our dose–escalation approach in OS revealed the central regulatory role of GPX4. This aligns with the foundational studies by Yang et al. and Friedmann Angeli et al. that established GPX4 inhibition as a sufficient trigger of ferroptosis (Friedmann Angeli et al., 2014; Yang et al., 2014). However, our mechanism diverges from classical GPX4 inhibitors such as RSL3 (a direct covalent inhibitor) or erastin (which depletes GSH, the essential cofactor for GPX4). Instead, high-dose PAL induces sustained oxidative stress that leads to NRF2 pathway inactivation and subsequent GPX4 protein degradation, representing a distinct, indirect route to disable the GPX4-dependent ferroptosis defense system. While Li Jiang’s work highlighted NO-mediated lysosomal dysfunction in PAL-induced ferroptosis using Ringer’s solution (Jiang et al., 2021), our findings establish the NRF2/GPX4 axis as a critical pathway in medium-activated PAL systems. These comparative insights underscore the context-dependent mechanism of PAL while expanding its therapeutic applicability.
Although there is concern that low-dose PAL activates the antiferroptosis pathway in OS cells, this issue does not undermine the findings presented here. The focus of this study was to investigate the impact of PAL dosage on OS cells under a fixed duration of PAL exposure. Both the results of this study and those from numerous other investigations (Bauer, 2019; Dai et al., 2023; Jo et al., 2022) demonstrate that the effects of PAL on OS cells are long-lasting and stable. As antioxidant factors are gradually depleted and the PAL exposure time increases, OS cells inevitably progress toward cell death. Consequently, as long as the PAL concentration reaches the threshold required to trigger oxidative defense mechanisms in cancer cells, PAL ultimately achieves an anticancer effect (Wang et al., 2024).
The potential of PAL therapy in clinical OS treatment lies in its ability to overcome chemoresistance: a major limitation of current regimens (Jo et al., 2024). The RONS produced by PAL selectively target tumor cells due to their inherently elevated oxidative stress, sparing normal tissues. This specificity, combined with the liquid state of PAL, enables deep-tissue penetration for targeting pulmonary metastases or residual postsurgical microtumors. While pulmonary metastasis involves systemic dissemination, our in vitro models focus on the initial local invasion and intravasation stages, which are prerequisites for distant metastasis. The inhibition of these processes by PAL implies a broader antimetastatic effect, potentially reducing the risk of pulmonary spread in vivo, as supported by studies in other cancers (Cai et al., 2023). Clinically, PAL could be deployed as an intraoperative irrigant during limb-salvage procedures or administered via precision intratumoral injections for unresectable lesions (Yao et al., 2025).
Translation to human applications faces three primary challenges. First, delivery optimization requires standardization of PAL formulations across biocompatible solutions such as Ringer’s lactate and saline, coupled with rigorous stability protocols to maintain reactive species efficacy during storage and administration. One promising approach involves integrating PAL with injectable hydrogels, which form depots at tumor sites for sustained release. This hydrogel–PAL synergy significantly prolongs reactive species exposure, addressing rapid RONS decay, while enabling precise spatial control to enhance therapeutic efficacy and reduce dosing frequency (Cao et al., 2025; Labay et al., 2020). Second, while our data demonstrate that high-dose PAL selectively induces ferroptosis in OS cells, we acknowledge concerns regarding potential unintended effects on normal cells. Our expanded in vitro assessment across multiple normal cell lines (hFOB, BMSCs, HIEC-6) revealed no significant impact on viability or disruption of the NRF2/GPX4 axis, supporting PAL’s selectivity likely due to elevated oxidative stress in tumor. However, comprehensive safety profiling must address potential systemic effects beyond our observed absence of hepatic/renal toxicity, including long-term immunomodulatory consequences and off-target tissue interactions, and we recognize that in vitro models cannot fully replicate the in vivo microenvironment. Therefore, future studies should prioritize in vivo models to evaluate off-target effects and ensure therapeutic safety. Third, dosing precision necessitates biomarker-guided titration—monitoring GPX4 suppression or lipid peroxidation markers—to establish tumor-specific therapeutic thresholds while avoiding paradoxical antioxidant activation (Yao et al., 2025). Furthermore, as highlighted in prior studies (Tanaka et al., 2016), the limited stability and penetration depth of plasma-derived reactive species necessitate innovative delivery strategies for deep-seated or systemic tumors. In our study, PAL is primarily targeted at residual neoplastic osteoblasts and micrometastases postresection, addressing chemoresistance-driven relapse as noted in the Introduction section. Compared with radiolyzed or ozonated water, PAL’s RONS repertoire is uniquely generated through CAP-induced reactions, leading to sustained oxidative stress that may be more effective in triggering iron-dependent cell death. Future efforts should focus on optimizing PAL for local applications, such as intraoperative irrigation to eradicate residual cells or intratumoral injection for accessible lesions, to minimize systemic toxicity. In addition, combining PAL with existing therapies (e.g., chemotherapy or immunotherapy) could synergize to overcome chemoresistance, as suggested by recent studies in other cancer models.
Compared with conventional ferroptosis inducers such as erastin and sulfasalazine, PAL offers distinct advantages: its multimodal redox modulation bypasses transporter-dependent uptake mechanisms that frequently underlie chemoresistance (Hassannia et al., 2019) while simultaneously generating immunogenic cell death signatures that may synergize with emerging checkpoint inhibitors (Miebach et al., 2023). Future studies should explore this combinatorial potential alongside technical innovations in PAL delivery systems for deep-seated tumors.
In summary, this was a pioneering study of the mechanism by which PAL exerts dose-dependent effects on the NRF2/GPX4 regulatory axis (Fig. 8), establishing ferroptosis induction through GPX4 suppression as a key antitumor mechanism in OS. The successful in vitro-to-in vivo translation validates the ability of PAL to overcome chemoresistance—a persistent clinical challenge (Hassannia et al., 2019; Niu et al., 2022)—by leveraging ferroptosis vulnerabilities. Our work not only elucidates the unique mode of action of PAL among CAP derivatives but also provides a rationale for optimizing PAL therapies through dose modulation.
Materials and Methods
Electronic laboratory notebook was not used
Ethics statement animal experiments
All animal experiments were approved by the Institutional Animal Care and Use Committee of Hefei Comprehensive National Science Center (No. IHM-AP-2024-024) and conducted in compliance with ARRIVE guidelines 2.0 (Supplementary Data). Experimental protocols followed the NIH Guide for the Care and Use of Laboratory Animals (8th ed.), with statistical power analysis determining sample sizes to minimize animal use. Randomization and blinded assessment procedures were implemented throughout the study.
Cell culture and reagents
Human OS cells (SAOS-2 and U2OS), hFOB, human BMSC, and HIEC-6 were purchased from Pricella Life Science & Technology Co., Ltd (Wuhan, China). All cells were cultured in Dulbecco’s modified Eagle’s medium (DMEM, Bio-channel, Nanjing, China) supplemented with 10% fetal bovine serum (Wisent, Nanjing, China) and 1% penicillin/streptomycin (New Cell & Molecular Biotech, Suzhou, China). The cells were maintained in a humidified incubator under 5% CO2 at 37°C and routinely checked for mycoplasma contamination.
Fe-1 (Cat. #347174-05-4) was purchased from Selleck Chemicals (Houston, TX, USA). NAC (Cat. #HY-B0215), carboxy-PTIO (Cat. #HY-18734A), and DFO mesylate (Cat. #HY-B0988) were purchased from MedChemExpress (Shanghai, China). RL solution was purchased from Macklin (Shanghai, China).
Plasma sources and preparation of PAL
A custom-made CAP source (Supplementary Fig. S1) composed of a controlled power supply, a plasma generator, a ground lead, and a high-voltage electrode was used in this study. The high-voltage electrode comprised three equilateral triangular stainless-steel plates, each separated by 1.5 cm. The CAP generator was connected to a power supply with an output voltage of 12 V. CAP was generated at a voltage of 20 kV (peak-to-peak) with a frequency of 24 kHz, and the surrounding air was ionized to produce a CAP jet, which was sprayed onto the liquid surface 4 cm away from the electrode sheet.
In in vitro cell experiments, prolonged contact between PAL and cells is essential; therefore, we selected complete DMEM as the liquid activation medium for CAP. Given the potential effects of serum and antibiotics in complete medium on immunodeficient nude mice, RL solution was chosen as the liquid medium for CAP activation in the in vivo experiments.
A total of 20 mL of untreated liquid was placed in a 70-mm-diameter crystallization dish. The irradiation time of CAP was differed (Fig. 1A), yielding five doses of PAL. The prepared PAL was used immediately after preparation. The CAP device reirradiated 20 mL of untreated liquid to ensure that the liquid in the Petri dish remained at 20 mL during the irradiation process.
In the context of plasma medicine, “PAL dosage” refers specifically to the duration of CAP activation time (in minutes), which serves as an established proxy for the cumulative reactive species load generated in the liquid phase (Bruggeman et al., 2016; Yan et al., 2015). This terminology convention is widely adopted in the field to quantify plasma treatment intensity, rather than denoting the concentration of a specific molecular compound.
Cell viability assay
In these studies, 1 × 104 cells/well were plated in 96-well plates. After a 12-h incubation period, the cells were treated with various doses of PAL for 12 or 24 h. Cell viability was assessed by the cell counting kit-8 (CCK-8) (Cat. #C0037, Beyotime Biotechnology, Shanghai, China) according to the manufacturer’s protocol. After incubation at 37°C for 1 h, the absorbance was measured with an optical density of 450 nm using a microplate reader (Thermo Fisher Scientific, Rockford, IL, USA).
In the experiment studying the effects of ROS scavengers and ferroptosis inhibitors on the tumor-killing efficacy of PAL, 4 h before PAL treatment, cells were pretreated with NAC (2 mM), Fe-1 (5 μM), DFO (1 mM), or carboxy-PTIO (200 μM) diluted in complete DMEM for 4 h. Then, the OS cells were treated with PAL15min for 12 h. Subsequently, the changes in cell viability in each group were observed according to the CCK-8 experimental procedure described above.
ROS measurement
Intracellular ROS were measured with the fluorescent probe DCFH-DA (Cat. #S0033S, Beyotime Biotechnology) according to the manufacturer’s protocol. Briefly, cells were incubated in 12-well plates (50,000 cells/well) with PAL15min or PAL20min at 37°C. At 12 h after PAL treatment, the cells were washed with PBS once and stained with the DCFH-DA solution (10 μM) for 30 min at 37°C in dark and then washed three times with PBS. Fluorescence was determined with a flow cytometer (FACSCanto BD Biosciences, Bedford, MA, USA).
Cloning formation assay
The OS cells were pretreated for 12 h using three different PAL doses. Following group allocation, cells were plated in six-well plates at 1 × 10³ cells/well density to allow colony formation for 14 days. Postculture processing involved sequential steps: medium removal, 20-min fixation with 4% paraformaldehyde, 20-min staining with 0.5% crystal violet, three washes with PBS, and final documentation through white-background plate photography.
Scratch assay
OS cells (5 × 105 cells each well) were seeded into six-well plates and cultured until reaching nearly 100% confluency. Cells were pretreated with the three PAL doses for 12 h before creating uniform scratches using 200-μL sterile pipette tips. The damaged monolayers were then maintained in low-serum medium (1% FBS) for 12 and 24 h. Cellular migration patterns were monitored at both time points using a light microscope with 5× magnification.
Transwell assays
For invasion assays, matrix gel mixtures (6 μL gel + 54 μL serum-free DMEM) were polymerized in Transwell inserts at 37°C for 8 h. PAL-pretreated OS cells (three doses, 12-h exposure) and untreated OS cells were resuspended in serum-free DMEM and seeded into the upper chambers at 5 × 104 cells/well. The basal compartment received 10% serum-containing medium as the chemoattractant. After 36 h of incubation to allow cell migration, noninvasive cells remaining on the upper membrane surface were removed using cotton swabs. Subsequently, the membranes were fixed with 4% paraformaldehyde for 20 min, stained with 0.5% crystal violet for 20 min, and washed three times with PBS. Invaded cells in three random fields were captured and counted under a light microscope at 10× magnification.
Proteomic analysis
Total protein was extracted from untreated SAOS-2 cells and SAOS-2 cells treated with PAL10min for 12 h (three samples for each control group and PAL group); one portion was used for sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS-PAGE) after the determination of the concentration, and the other portion was digested with trypsin. Tryptic peptide mixtures labeled with tandem mass tags (Thermo Scientific, Rockford, IL, USA) were prepared according to a standard procedure. Six labeled peptide mixtures were combined and subjected to reversed-phase chromatographic separation and mass spectrometry analysis by Shanghai Luming Biological Technology. Database searching was conducted using Proteome Discover 2.4 (Thermo Fisher, San Jose, CA, USA) with the following parameters: static modification: tandem mass tag 6-plex (N-term, K); dynamic modification: oxidation (M), acetyl (N-term); digestion: trypsin; ms1 tolerance: 10 ppm; ms2 tolerance: 0.02 Da; missed cleavages: 2. Gene Ontology terms and Kyoto Encyclopedia of Genes and Genomes pathway analyses were used for biological categorization of the significantly differentially expressed proteins. Raw data were upload in iProX (Accession ID: IPX0009581000).
Western blot
Cells (2 × 105 cells per well) were evenly inoculated in each well of a six-well plate. After 24 h, cell confluence reached nearly 50%. At this point, the medium in each well was replaced with 2 mL of different doses of PAL. The cells were collected after 4, 8, or 12 h of PAL treatment. Cells were washed twice using ice-cold PBS and lysed in RIPA buffer (Cat. #P0013B, Beyotime Biotechnology) containing protease and phosphatase inhibitor cocktail (Cat. #P001, New Cell & Molecular Biotech). The protein concentration was quantified using a BCA protein assay kit (Cat. #WB6501, New Cell & Molecular Biotech). Equal amounts of protein extracts (50 μg) were subjected to SDS-PAGE, transferred onto polyvinyl difluoride (PVDF) membranes (Millipore, Billerica, MA, USA), and blocked with 5% nonfat dry milk for 1 h at room temperature. Subsequently, the membranes were incubated with primary antibodies at 4°C overnight. The primary antibodies used are listed in Supplementary Table S1. The membranes were then incubated with the appropriate horseradish peroxidase-conjugated secondary antibodies at room temperature for 1 h. After each step, the sections were washed with 1× TBST (1% Tween-20 in Tris-HCl buffer) seven times for 5 min each. Finally, the Western blots were developed using the Super ECL Detection Reagent (Cat. #36208ES60, Yeasen, Shanghai, China) and the expression levels of reactive protein bands were quantified using ImageJ 1.54f software.
Following the initial antibody detection, membranes were stripped to remove bound antibodies for reprobing. Briefly, the PVDF membranes were washed with 1× TBST for 15 min, followed by incubation with a commercial antibody stripping buffer (Cat. #PS107S; Epizyme, Shanghai, China) for 15 min with gentle agitation. The membranes were then thoroughly washed again with 1× TBST for 15 min. After stripping, the membranes were reblocked and subsequently reprobed with a different primary antibody, starting from the blocking step as described above. This protocol allowed for the sequential detection of multiple targets on the same membrane.
Immunofluorescence using confocal microscopy
All fluorescence experiments in this study were performed using cells seeded on 35 mm confocal dishes (Cat. #BS-15-GJM; biosharp, Shanghai, China). Specifically, 150,000 OS cells were seeded on these dishes and allowed to adhere for 24 h under standard culture conditions to ensure a stable cell state before initiating PAL treatment. After OS cells were treated with PAL10min for 12 h, the cells were fixed with 4% (w/v) paraformaldehyde for 10 min/RT, washed three times with PBS, incubated with 0.1% (v/v) Triton X-100 and blocked for 1 h at RT with 3% (w/v) bovine serum albumin. The cells were then incubated overnight at 4°C with primary antibodies against NRF2 (Cat. #16396-1-AP; 1/200 dilution; Proteintech, Wuhan, China). The following day, the sections were washed three times with PBS (5 min per wash) and then incubated with Alexa Fluor 488-labeled goat anti-rabbit IgG (H + L) (Cat. #A0423, Beyotime Biotechnology) as a secondary antibody. Antifade mounting medium with DAPI (Cat. #P0131, Beyotime Biotechnology) was added dropwise onto the samples, and the sections were then covered with glass coverslips. Images were obtained using a confocal microscope (LSM 980, Carl Zeiss, German).
Detection of intracellular ferrous ion content
A FerroOrange fluorescent probe (Cat. #F374, Dojindo, Japan) was used according to the manufacturer’s protocol for the detection of intracellular Fe2+. In brief, after OS cells were treated with PAL20min or DFO mesylate (1 mM) for 12 h, the cells were cultured with 1 μM FerroOrange for 30 min and observed under a confocal microscope (Ex/Em: 561 nm/570–620 nm). To facilitate observation and analysis, the fluorescence signal from FerroOrange was marked as orange. Cell images were taken using a laser confocal microscope (LSM 980, Carl Zeiss).
Assessment of lipid peroxidation
After treating OS cells with PAL20min or Fe-1 (5 μM) for 12 h, the cells were subsequently incubated with a 50 μM C11-BODIPY™ 581/591 probe (Cat. L267, Dojindo) away from light for 1 h. Cells were washed twice with PBS to remove the excess probe, and then a small amount of serum-free medium was added to cover the cells in the culture dish. Cell images were taken using a laser confocal microscope (LSM 980, Carl Zeiss).
Lipid MDA assay
OS cells were treated with PAL20min for 12 h. MDA levels were measured using an MDA Assay Kit (Cat. #M496, Dojindo). The reaction mixture was evaluated on a fluorescence microplate reader (Thermo Fisher Scientific, USA), with MDA levels expressed as nmol/mg protein.
RNA isolation and RT-qPCR
Total RNA was isolated from OS cells using a TRIzol RNA Kit (Cat. #M5102, New Cell & Molecular Biotech) according to the manufacturer’s instructions. Hifair® V Reverse Transcriptase (Cat. #11300ES92, Yeasen) was used for reverse transcription. The cDNA was generated from mRNA using Hieff® qPCR SYBR Green Master Mix (Cat. #111202ES08, Yeasen). RT-qPCR was then performed using TB Green on a QuantStudio 3 (Thermo fisher scientific, USA). The primers used are listed in Supplementary Table S2. 18S RNA was used as the endogenous control.
Transfection of FSP1, GPX4, and NRF2
The sequences of the FSP1, GPX4, and NRF2 overexpression plasmid and small interfering RNAs (siRNAs) (Supplementary Table S3) were synthesized by HANBIO (Shanghai, China), and the lyophilized constructs were dissolved in DEPC-treated water at 20 μM. Plasmids for FSP1, GPX4, and NRF2 overexpression were transfected into cells using LipoFiter 3.0 (HANBIO, China), and siRNAs were transfected into cells with CALNPTM RNAi (D-Nano Therapeutics Co., Ltd., Beijing, China). Briefly, the reagent and nucleic acid were dispersed in Opti-MEM and mixed. The solution was then added to cells and cultured in an incubator under 5% CO2 at 37°C for 48 h.
Mice
Twenty-four male BALB/c nude mice aged 4 weeks with an average weight of 16 ± 2 g were purchased from GemPharmatech (Nanjing, China) and randomly divided into four groups: PAL-pretreated (Pp), control, NC, and PAL. The Pp group received a 100 μL injection of the RL mixture containing 5 × 106 PAL20min-pretreated SAOS-2 cells, which had been pretreated for 12 h, in the left axilla. The remaining three groups received a 100 μL injection of the RL mixture containing 5 × 106 untreated SAOS-2 cells at the same location. Three weeks after cell inoculation, the volume of subcutaneous tumors in mice in the control, NC, and PAL groups was ∼100 mm³, at which point intervention treatments were initiated. Nude mice in the NC and PAL groups received intratumoral injections of 100 μL of RL and 100 μL of PAL20min, respectively. The volume of subcutaneous tumors was measured and recorded before each intratumoral injection. Nude mice in the control and Pp groups did not undergo any intervention during the treatment period. The treatment was repeated every other day until receiving a total of seven treatments. In the subsequent OS xenograft models, animals were randomized into the PAL and PAL + Fer-1 groups following the experimental scheme in Figure 7A, with the PAL group receiving intratumoral injections of 100 μL PAL20min liquid and the PAL+Fer-1 group receiving 100 μL PAL20min liquid containing 5 μM Fer-1.
Mice were anesthetized with ketamine (10 mg/mL) intraperitoneally before treatment; the injection volume was 10 μL/g of mouse body weight. The tumor volume was calculated using the following formula: volume = length × width2 × 0.5. Tumors were dissected after the mice were sacrificed on the 35th day, with day 1 being when the mice were inoculated with tumor cells.
Immunohistochemistry
For immunohistochemistry, the primary antibodies used were anti-4-HNE (1:200 dilution, Cat. #ab46545, Abcam) and anti-GPX4 (1:200 dilution, Cat. T56959M, Abmart). In brief, the serial sections were deparaffinized with xylene and ethanol. Samples were immersed in Tris-EDTA solution (pH 8.0) and incubated with boiled water for 20 min. The sections were incubated with blocking buffer (5% BSA in 1× PBS) for 30 min at room temperature and then incubated with a primary antibody overnight at 4°C. The antibodies were detected using a Peroxidase/DAB kit (Cat. #PV-9000, ZSGB-BIO, Shanghai, China). Following fixation, the tissue sections were observed at 20× magnification. Positive staining (%) was calculated using ImageJ 1.54f software. The staining ratio (%) was calculated as (Positive area/Total area) × 100.
Statistical analyses
All statistical analyses were conducted using GraphPad Prism 9.5 software (GraphPad Software, La Jolla, CA). Data are presented as mean ± standard deviation. The significance of differences was assessed through unpaired t-tests, one-way ANOVA, or two-way ANOVA followed by Tukey’s multiple comparison test. A p value of <0.05 was deemed statistically significant.
For fluorescence intensity quantification, images were analyzed using ImageJ software. A consistent threshold was applied to define regions of interest (ROIs) based on signal-to-background ratios. The mean fluorescence intensity (MFI) was calculated by dividing the total fluorescence intensity by the pixel area of the ROI. MFI values were normalized to the average MFI of the control group from three independent replicates.
Authors’ Contributions
L.X., M.Z., and P.H. conceived and designed the experiments and wrote the article. L.X., M.Z., H.W., and S.L. performed the experiments. L.X., X.S., and Y.H. coordinated and supervised the work. L.X. analyzed and interpreted the data. Y.W. designed and provided the plasma source. All authors read and approved the final article.
Footnotes
Acknowledgments
The authors extend their sincere gratitude to the Center for Scientific Research and the Institute of Clinical Pharmacology at Anhui Medical University for their provision of numerous instruments and a high-quality experimental environment. Graphical abstract, 1A, 7A, and 8 were created, in part, with ![]() .
.
Author Disclosure Statement
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this article.
Funding Information
This study was funded by the Program for Upgrading Basic and Clinical Collaborative Research of Anhui Medical University (grant number: 2020xkjT033), Major scientific research projects of the Health Commission of Anhui Province (grant number: AHWJ2023A10008), and Program for Upgrading Basic and Clinical Collaborative Research of Anhui Medical University (grant number: 2023xkjT031).
Disclaimer
The authors confirm that this article has not been published and is not under consideration for publication elsewhere. All the authors have agreed with the present submitted version of this article.
Supplemental Material
Abbreviations
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.