
Abstract
Amyotrophic lateral sclerosis (ALS) is a neurodegenerative condition noteworthy for upper and lower motor neuron death. Involvement of respiratory motor neuron pools leads to progressive pathology. These impairments include decreases in neural activation and muscle coordination, progressive airway obstruction, weakened airway defenses, restrictive lung disease, increased risk of pulmonary infections, and weakness and atrophy of respiratory muscles. These neural, airway, pulmonary, and neuromuscular changes deteriorate integrated respiratory-related functions including sleep, cough, swallowing, and breathing. Ultimately, respiratory complications account for a large portion of morbidity and mortality in ALS. This state-of-the-art review highlights applications of respiratory therapies for ALS, including lung volume recruitment, mechanical insufflation-exsufflation, non-invasive ventilation, and respiratory strength training. Therapeutic acute intermittent hypoxia, an emerging therapeutic tool for inducing respiratory plasticity will also be introduced. A focus on emerging evidence and future work underscores the common goal to continue to improve survival for patients living with ALS.
Keywords
Introduction
Amyotrophic lateral sclerosis (ALS) is a neurodegenerative disorder characterized by progressive upper and lower motor neuron death leading to worsening atrophy and weakness of the limbs, trunk, and bulbar muscles. 1 Whether occurring sporadically or in association with a known genetic predisposition, ALS most commonly presents as spinal-onset characterized by atrophy or weakness of one or more limbs or trunk, or bulbar-onset disease, marked by decrements in bulbar muscle coordination that alter the patency of the upper airways and deteriorate airway protective behaviors, such as cough and swallowing. 2 In contrast to spinal- and bulbar-onset sporadic phenotypes, which represent ∼95% of cases, ALS rarely presents as a purely respiratory onset (<5%). 3 However, people with ALS inevitably encounter respiratory muscle weakness within the disease.2,4
Respiratory motor neuron death can induce denervation and partial reinnervation of affected respiratory muscle fibers by adjacent motor axons (Figure 1). Collateral reinnervation results in larger, composite motor units that can preserve motor function,
5
but may alter complex activities such as management of sialorrhea and breathing-swallowing coordination.
6
Airway involvement in ALS may include progressive airway obstruction
7
and velopharyngeal insufficiency,
8
leading to ineffective airway protection and risk for aspiration. Loss of respiratory motor neurons leads to weakness and atrophy of the respiratory muscles and progressive hypoventilation. Ultimately, chronic respiratory failure leads to shortened survival.9,10 Respiratory upper and lower motor neuron degeneration in ALS leads to weakness and atrophy of the inspiratory and expiratory muscles, as well as muscles of the upper airway. Isolated or combined dysfunction of these motor neuron pools leads to hypoventilation, dystussia, and dysphagia in patients.
Two ALS disease-modifying drugs, riluzole and edaravone, are FDA-approved in the US, 11 while a third medication, sodium phenylbutyrate/taurursodiol recently received approval in the US conditional approval in Canada. 12 Each of these medications aim to slow disease progression, improve quality of life, and enhance survival.11,13,14 In conjunction with pharmacological approaches, early detection and management of respiratory function and airway protection are crucial to preserving function in ALS and prolonging survival. 15 Patient-centered management of respiratory compromise requires knowledge and expertise in the use of airway clearance assistance, invasive or noninvasive mechanical ventilation, dysphagia management and oral care 16 and rehabilitation techniques to improve respiratory neuromuscular function.
This state of the art review summarizes relevant literature pertaining to the effects of respiratory therapies in ALS. The existing and emerging therapies highlighted in the review include mechanical insufflation-exsufflation (MI-E), lung volume recruitment (LVR), non-invasive ventilation (NIV), respiratory strength training exercises (RST), and acute intermittent hypoxia (AIH). A symbol (♦) indicates evidence to suggest one of these therapies may support ventilation, cough, or swallow function. Since much of the existing research consists of small cohort or retrospective studies with some risk of bias, results should be interpreted with caution and indicate further controlled studies are needed.

Airway clearance
Cough is an important protective mechanism to clear excess secretions, irritants, and foreign matter from the airways.17,18 As illustrated in Figure 2, generation of adequate cough requires a complex coordination of the inspiratory muscles for a deep inspiration, increasing lung volume, upper airway muscles for rapid, brief glottic closure and vigorous expiratory muscle contraction to increase intrathoracic pressure. Lastly, the abrupt opening of the glottis expels material from the airways. Impaired timing and coordination of any of these phases can lead to dystussia in ALS (Figure 1).
19
Bulbar dysfunction reduces the rapid opening and closing of the glottis during cough, even when respiratory strength remains within normal limits.
20
Examples of cough spirometry waveforms in an unaffected individual (a) and a person with ALS (b). a) The example from the unaffected individual illustrates clear temporal distinctions between the inspiratory, compression, and expiratory phases of cough. An increased peak inspiratory flow reflects an elevated inspiratory effort to augment the operating volume of the lungs. Peak cough flow (PCF) refers to the peak flow achieved during the expiratory phase of cough and is normal in this example (6.5 L/s). b) The voluntary cough waveform in the ALS subject demonstrates prolonged inspiratory and compression phases, presence of air leaking during compression phase, prolonged time to reach peak cough flow, and a lower absolute expiratory peak flow, which contributed to an ineffective cough (<3 L/s).
Peak cough flow (PCF) is the greatest expiratory airflow achieved during the expulsive phase of cough and is essential for both airway patency and mucus clearance. An effective cough usually requires a PCF greater than 162 L/min, 21 and normal PCF typically exceeds 360 L/min. 22 Expiratory muscle weakness reduces expiratory airflow generation, leading to decreased cough efficacy and secretion clearance. 23 Poor airway clearance may lead to atelectasis, acute respiratory insufficiency, and pneumonia. Ineffective airway clearance and retained secretions may lead to airway obstruction, atelectasis, acute respiratory insufficiency, and infections, which increase the work of breathing.17,18
Mechanical insufflation-exsufflation
Mechanical insufflation‐exsufflation (MI‐E) is accomplished with a purpose-built device that uses positive pressure to deliver external inflation of the lungs; after a brief pause, inflation then rapidly transitions to negative pressure, reproducing high airflow rates typically generated during the expiratory phase of cough (Figure 3). The goal of MI-E is to create an expiratory flow with sufficient velocity to overcome shear forces in the airway, to mobilize secretions from the lower airways for removal, and assist with sputum clearance in those with insufficient voluntary cough force.
23
Thus MI-E directly impacts dystussia and may improve ventilation and perceived work of breathing (Table 1). Schematic illustrates MI-E pressure and timing, as compared to bilevel, positive pressure ventilation. A single MI-E set alternates positive insufflation pressure and brief inspiratory hold, with rapid application of negative exsufflation pressure. The inspiratory and expiratory holds, along with pauses between I-E cycles, can be customized to correspond to the patient’s spontaneous breath timing. I-E cycles are frequently repeated a few times in a single I-E set, and multiple I-E sets are typically performed in a single assisted airway clearance session.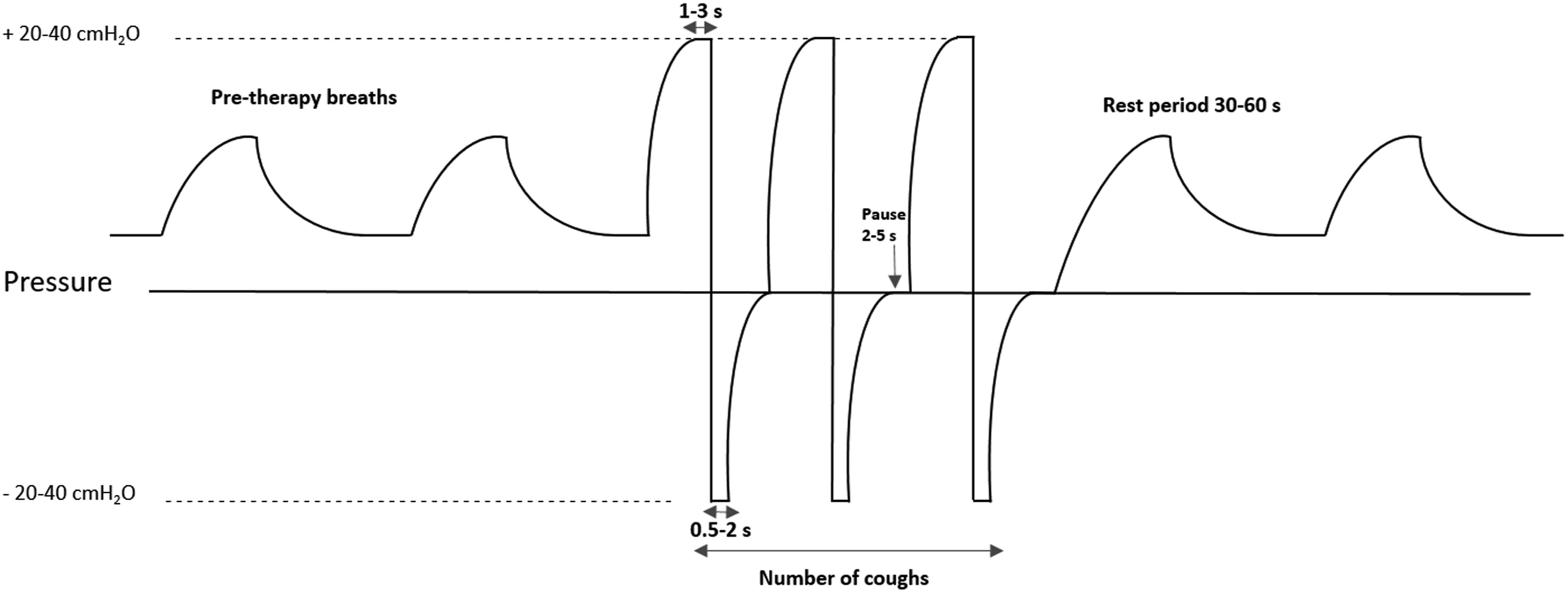
The “dose” and timing of external pressures and airflow rates are essential to patient tolerance of MI-E equipment. The high external pressures require a closed circuit with tightly sealed mask or direct connection to a tracheostomy tube. Manufacturers generally recommend titrated pressures ±40 cm H2O for both inspiration and expiration. However, patients with bulbar dysfunction may experience upper airway collapse during MI-E and could require lower pressure settings, especially during insufflation. 24 The applied airway pressures of MI-E may also require reduction for low BMI, short stature, or restrictive lung disease, to reduce the risk of barotrauma or pneumothorax. 25 A recent practice analysis noted that, in those with neuromuscular disease, lower pressures and shorter times were more used for insufflation than exsufflation. 26 While some patients may be able to independently place the circuit, most typically require assistance from a trained caregiver, and functionally dependent patients may be able to tolerate well-titrated, passive MI-E delivery. 26
MI-E can be a useful tool in managing airway patency and helpful for ALS patients who are ventilator dependent. For example, in a small study of ventilator-dependent patients with ALS, MI‐E was more effective in removing respiratory secretions and alleviating work of breathing, compared to airway suctioning. Patient comfort and perceived efficacy were also greater with MI-E. 27 Several studies demonstrate that MI-E acutely augments voluntary PCF in a variety of neuromuscular diseases (NMDs).28–30 Another study of MI-E in ALS and various other NMDs reported acutely enhanced forced vital capacity (FVC), a common prognosticator for respiratory failure, for at least 1 h following the treatment. If MI-E can help improve airway patency, it may improve and optimize airway function for patients. However, the magnitude of the improvement varied between different NMDs. 31
In a practice assessment of MI-E in NMD, Chatwin and Simonds, observed patients with tracheostomy and those with larger quantities of secretions were more likely to incorporate MI-E intro their pulmonary hygiene routine. The authors emphasized the importance of comprehensive patient and caregiver education to increase adherence to MI-E. 26 Patients with ALS perceive that MI-E optimized their secretion clearance and respiratory symptoms, when patients trusted their caregivers, and when caregivers felt confident in the MI-E procedure and equipment. 32 A retrospective analysis reported that addition of daily MI-E to routine pulmonary hygiene may significantly increase survival. In those who did not use external ventilatory support, the addition MI-E did not prolong survival. In contrast, the addition of at least one daily MI-E session to a regimen of bilevel positive pressure ventilation extended survival an average of 10 months. However, the survival benefit was only observed in those without bulbar dysfunction, and the study was restricted to a single center in the US. 33
Recent developments
Recent work by Steindor evaluated the acute effect of MI-E on small airway function, using the lung clearance index (LCI). 34 LCI is calculated by measuring changes in concentration of a tracer gas during resting breathing, with higher LCI values reflecting greater small airway disease and ventilation inhomogeneity. 35 Compared to controls, LCI was increased in those with NMD. These results reveal that pathological lung involvement in NMD extends beyond pulmonary restriction secondary to neuromuscular weakness, although the clinical significance of the results remains unclear. Further, a higher LCI corresponded with lower FVC and PCF, indicating greater pulmonary involvement with progressive weakness. The use of MI-E did not yield immediate improvements in LCI, though individuals with ALS were not specifically studied in this cohort. 34
One consideration for bulbar patients using MI-E is to plan a patient-centered protocol to address any airway difficulties they may be experiencing. Patients with bulbar dysfunction require particular attention to identify MI-E settings that do not trigger vocal fold adduction or laryngeal muscle spasms that can restrict airflow. 36 A recent analysis of flow-time waveforms during MI-E aimed to optimize pressure settings in a cohort of individuals with ALS. Obstruction during insufflation occurred in nearly 50% and was associated with upper motor neuron bulbar disease. In contrast, airway collapse during exsufflation was identified in 26% and corresponded with bulbar lower motor neuron involvement. Adjustments in insufflation and/or exsufflation pressure parameters either upper or motor neuron involvement helped to resolve the upper airway obstruction during MI-E. The authors noted that most adjustments led to lower insufflation pressures than exsufflation, 37 which is consistent with other reports. 26
Future studies
Airflow-pressure ramping algorithms are proprietary and differ between devices. 38 In a recent case report, the use of a breathing simulator identified pressure-timing differences between MI-E machines that improved both patient comfort and cough flows. 39 Controlled studies are needed to evaluate whether device changes more broadly improve MI-E tolerance when the treatment fails. While MI-E studies often cite PCF as a dependent variable, prospective, controlled trials are needed to evaluate additive effects of MI-E versus usual care on PCF as well as other clinical outcomes, including impact alveolar ventilation, gas exchange, secretion clearance, and breathing perception. One retrospective study reported incidental findings on the effects of differing regimens of noninvasive ventilation and MI-E on survival. 33 Further prospective research is needed to evaluate longer-term effects of MI-E on preserving quality of life and reducing respiratory morbidity and mortality in ALS.
Dysphagia
Swallowing is a preventive airway protective behavior that consists of complex, highly coordinated actions. It entails oral, oropharyngeal, and esophageal phases that must be well-timed with breathing and requires more than 30 muscles to execute effectively. Unfortunately, over 80% of patients living with ALS will experience dysphagia, or impaired swallowing. 40 Patients with bulbar onset report early difficulties with speech, mastication, and swallowing. With spinal-onset, patients may eventually develop dysphagia with disease progression. Dysphagia can emerge in ALS due to a host of deleterious factors (i.e., restricted tongue movement, laryngeal dysfunction, tongue atrophy, poor bolus control, etc.) that may be caused by the degeneration of cortical motor neurons, corticobulbar tracts, and brainstem nuclei.41,42 Patients may also experience loss of sensation, which can lead to silent aspiration. 43 The vast heterogeneity of ALS may lead to various degrees of facial, lingual, pharyngeal, and laryngeal dysfunction, as well as muscle weakness, stiffness, and/or spasticity. These impairments inevitably affect the motility, strength, and coordination of muscles within the oral cavity, pharynx, and larynx. Discoordination and weakness may also result in residues of food or mucosal secretions in the airways, often indicating the oral phase of swallowing is impacted first. 41
Maintenance of airway patency and proper nutrition are paramount to clinically manage ALS. Dysphagia and impaired cough, or dystussia, can antagonize these goals. Additionally, the ability to eject foreign materials from the airways with a productive cough is necessary for a safe swallow but is often impaired by respiratory muscle weakness. 44 Complications of dysphagia and impaired cough can lead to malnutrition, dehydration, aspiration pneumonia, respiratory insufficiency, and decreased medication compliance, that may ultimately contribute to social isolation and lower quality of life.45,46 While regular monitoring of swallow and cough function is imperative, subtle changes in swallow function can be difficult to detect, and standardized dysphagia management protocols in ALS are lacking. Early, regular dysphagia evaluations are necessary to prevent complications, particularly if patients cannot sense difficulties (i.e., aspirations, increased mealtimes, fasciculations, etc.). 40
Collaboration between speech language pathologists and neurologists can optimize diagnosis and management of dysphagia in ALS. Patient attendance at a multidisciplinary clinic is also recommended for monitoring of appropriate caloric intake, respiratory function, and cough efficacy. Imaging tests for dysphagia and aspiration include the modified barium swallow study (MBSS) and fiberoptic endoscopic evaluation of swallowing (FEES), which are considered gold standards to assess swallow function. 45 The Revised ALS—Functional Rating Scale (ALSFRS-R) is a validated survey that measures daily function in ALS. It is the only measure consistently administered and used to evaluate and monitor progression of overall disability, including dysphagia, in the clinical setting. 47 Higher ALSFRS-R scores are reported in patients that demonstrate a safe and effective swallow, and the ALSFRS-R can be used to track dysphagia. 48 Other surveys and assessments of swallowing include the 3-ounce water challenge, the Penetration Aspiration Scale (PAS), the Eating Assessment Tool (EAT-10), and the Functional Oral Intake Scale (FOIS). Dysphagia evaluations serve multiple purposes: 1) to assess swallow function, 2) monitor progression of ALS bulbar symptoms, and 3) inform providers on whether percutaneous endoscopic gastrostomy (PEG) or radiologically inserted gastrostomy (RIG) may be indicated. 16
Although patient-centered care of dysphagia is a primary goal, standardized protocols are lacking for clinicians, and the progressive nature of ALS limits the available options. However, clinicians should begin by providing educational resources regarding the anatomy and physiology of the swallowing mechanism. Speech language pathologists can also suggest: modifying diets using the international Dysphagia Diet Standardisation Initiative (IDDSI) framework, postural compensation maneuvers to preserve oral feeding (i.e., chin tuck, trunk positioning), good oral hygiene practices (i.e., suctioning toothbrush), specialized utensils to assist with bolus delivery, modified eating practices (i.e., small sips, small bites, remove straws, etc.), and pleasure feeds to improve quality of life and increase social interactions. A PEG is typically recommended when weight loss exceeds 10% of the patient’s pre-diagnosis weight and FVC <50% of the predicted value (or before SNIP <40 cm H2O).49,50 If patients exceed these thresholds, or if patients have bulbar onset, RIG may be recommended. Patients with PEG or RIG may still take food by mouth or engage in pleasure feeds and receive nutritional intake via the PEG or RIG. 51 Should patients experience poor bolus control, PEG or RIG may be considered to maintain and monitor appropriate caloric intake.
Lung volume recruitment
Lung volume recruitment (LVR) is a procedure consisting of multiple sequential inhalations and glottis closures (also referred to as “breath stacking”), with the objective to produce lung inflation beyond a patient’s single spontaneous inspiratory efforts. Breaths may be stacked with voluntary efforts, or assisted by an “ambu” style bag or a positive pressure mouthpiece ventilation. The assisted inflation is typically followed by a breath hold and slow exhalation to recruit under-expanded alveoli. Alternatively, the extra inspiratory volume can also be channeled into a forced expiration, to improve cough force (Table 1).52,53 The elastic recoil pressure of the lungs increases at higher operating volumes; this recoil then aids force generation during voluntary expiratory efforts to improve PCF.54,55
Respiratory muscle weakness erodes vital capacity over time and may also lead to increased stiffness of the lungs and chest wall. Chronic atelectasis and airflow limitations may also increase susceptibility to respiratory infection, particularly in chronic neuromuscular disease. 56 Several studies undertaken in a multitude of neuromuscular conditions, reveal LVR may acutely increase maximum insufflation capacities 53,55,57–59 and PCF.19,24,29 Breath-stacking can lead to immediate, short-term improvements in lung function 31 without altering voluntary inspiratory strength, 58 and may lead to a transient improvement in respiratory system compliance. 55 For up to 30 min post-LVR, significant improvements in PCF and the efficiency of airway protective strategies, such as throat clearing, may be observed; which may reduce the risk of aspiration during meals. 60 Repeated, daily use may promote improved respiratory mechanics and enable patients with neuromuscular conditions to preserve their insufflation capacity. 58 The timing of LVR initiation matters. Patients who begin using LVR when PCF is already below the critical threshold of 160 L/min may yield a limited clinical benefit on cough efficacy. 61
While some work suggests that LVR may help preserve pulmonary function over time,17,53 only one controlled study has reported the effects of LVR on voluntary cough clinical outcomes of patients with ALS. 62 After randomization, patients underwent similar repetitions of either LVR or MI-E therapy, twice daily, for up to 12 months. This controlled study demonstrated no differences after 12 months of use between breath stacking and MI-E on caretaker strain, hospitalization for respiratory infection, quality of life, or survival, but the LVR equipment was more cost-effective. Conclusions were limited by sample variability that limited statistical power. 62
Recent developments
To monitor compliance with LVR, Naughton and colleagues 63 designed and assessed an “LVR counter” that recorded bag compression during LVR sessions. The device was designed to fill a gap in assessing adherence to LVR sessions. The device could detect single squeezes of the LVR bag but lacked the ability to calculate volume or flow delivered. The ability to record compliance with prescribed LVR could be useful to clinically monitor patient tolerance or to assess feasibility of LVR in clinical research. The Naughton study confirmed patients could perform an adequate mouthpiece seal and compress the LVR bag twice daily, but the optimal “dose” of LVR remains to be determined.
Impacts of single LVR sessions on pulmonary function, voluntary cough, and respiratory compliance have been collectively studied in neuromuscular disease, but until recently LVR effects were not contrasted between ALS and slowly evolving conditions. Patients underwent two sets of five, individually titrated maximal inflations, and respiratory testing after the LVR session was compared to the pre-session baseline. Acute physiological effects in the sample included increases in average respiratory compliance, lung insufflation capacity, PCF, functional residual capacity, and total lung capacity. Subgroup analysis indicated that improved compliance effects were primarily influenced by improvements in the ALS group. 56
Future studies
While LVR transiently increases voluntary cough production, its effect on reflex cough is less understood and could impact a patient’s ability to detect and prevent aspiration. 43 Since LVR alone may not elicit a durable improvement in cough function, an ongoing multicenter clinical study (NCT03202017) 64 aims to investigate whether combinatorial LVR and expiratory strength training improve speech, breathing, and airway protection, measured through PCF, MEP, FVC, an eating assessment tool, swallowing related quality of life, and a speech intelligibility test.
Hypoventilation and ventilatory failure
The diaphragm is the primary inspiratory muscle, and dysfunction leads to decreased lung volumes, breathing dysfunction, dyspnea, reduced chest wall expansion, and alveolar hypoventilation. As inspiratory involvement builds, impaired airway clearance and hypoventilation emerge during the daytime. Consequently, chronic respiratory insufficiency and respiratory failure shorten survival.9,10 An early manifestation of respiratory involvement in ALS is sleep hypoventilation, which may be accompanied by obstructive or central sleep apnea.65–67 Respiratory muscle weakness and/or mechanical disadvantage in the recumbent position contribute to alveolar hypoventilation, during sleep. While an estimated 54 million US adults have clinically significant obstructive sleep apnea, 68 it is even more prevalent in people with ALS than the general population. 65
Sleep studies are essential for detection and treatment of sleep-related hypercapnia. The American Academy of Neurology’s (AAN) practice parameters for ALS indicate that nocturnal oximetry may be considered to detect hypoventilation, and non-invasive ventilation (NIV) should be considered at the earliest sign of either nocturnal hypoventilation or respiratory insufficiency (FVC <50% of predicted value) to prolong survival. 69 Although recent work found nocturnal hypercapnia predicts shorter survival of patients, early initiation of NIV can stabilize survival.65,70 Nevertheless, regular, ongoing assessment is required after initiation of nocturnal NIV; emergence of elevated CO2 or symptoms of daytime hypoventilation (e.g., dyspnea, fatigue, headache) indicate a need to increase ventilator settings and/or extend wearing schedule to include daytime hours.
Non-invasive ventilation (NIV)
Since hypoventilation contributes to significant morbidity and mortality in ALS, NIV remains a staple of preventative respiratory care for ALS (Table 1). Initial use may focus on correction of nocturnal dysfunction71,72 and expand to daytime support with disease progression. Devices can deliver bilevel, volume- or pressure-controlled breath delivery at specific times or coordinated with spontaneous efforts. Alternatively, devices may target a threshold ventilation through adjustable, volume-assured pressure support. Adaptive ventilator settings enable the device to detect and compensate for lapses in the baseline ventilation, despite progressive respiratory weakness. Patients may initially require slightly longer time to adjust to adaptive ventilator modes, adherence is similar over the longer term. 73 A variety of noninvasive interfaces (e.g., oronasal, nasal, mouthpiece) enable therapists to tailor support to suit other communication and swallowing needs. Moreover, some patients use their ventilator to take multiple simultaneous “stacked” breaths to increase their operating volume during voluntary coughing. 19
The seminal controlled trial of NIV in ALS randomized 92 patients with ALS and evidence of hypoventilation, to receive either NIV or standard supportive care. 74 Those with milder bulbar dysfunction used higher pressure settings and wore NIV more hours of the day; these patients reported an improved quality of life and survived a median 7 months longer than those assigned to standard care. However, >50% patients in the standard care group survived 11 days or less. 74 Similar benefits were not observed in those with poor bulbar function. Despite these limitations, subsequent clinical studies and retrospective chart reviews confirmed or extended the conclusions from the pivotal NIV trial. Multiple controlled trials illustrate that NIV can prolong survival, stabilize pulmonary function, reduce respiratory symptoms, and improve quality of life.15,75–78 Reported survival benefits range from 10 to more than 19 months 15,75,77,79,80 Early initiation of NIV, when vital capacity remains >80% predicted, provides a greater reduction of mortality risk than conventional prescribing guidelines and increases NIV adherence.15,72
At least 4 h of daily use appear to correspond to prolonged survival.15,80 A close assessment of sleep architecture in ALS demonstrates NIV can significantly ameliorate sleep-disordered breathing. 71 Improvements in sleep efficiency, proportion of time in deeper sleep and rapid eye movement stages, arousals, and gas levels were observed with NIV use. 81 Patient-reported benefits of NIV include improved quality of sleep, decreased daytime headaches, and decreased daytime fatigue. 82 Nevertheless, up to 40% of patients may not be able to tolerate four or more hours of regular use. 80
Efficacy of NIV may vary with ALS phenotype. In the pivotal study, NIV enhanced quality of life in those with poor bulbar function, but it did not extend survival. The authors observed that patients with severe bulbar involvement appeared to have less severe hypoventilation upon enrollment and recommended further study on approaches to improve wearing tolerance in this subset of patients. 74 Many15,72,80 but not all71,75 subsequent studies also reported lower efficacy of NIV for those with bulbar symptoms. With volume-assured pressure support modes of ventilation, fluctuating pressures can lead to greater airway collapse and residual apneas in bulbar disease. 73 While benefits of NIV may be inconsistent in those with bulbar dysfunction, safe implementation appears possible with carefully selected NIV settings and use of an oronasal mask. 83
Recent developments
NIV initiation guidelines from the American Academy of Neurology traditionally included maximal voluntary effort tests such as vital capacity16,69 which might not directly reflect resting ventilatory deficits or reflect sleep hypoventilation.84,85 European NIV prescribing criteria have been updated in recent years and offer additional options beyond pulmonary function and respiratory strength tests, including the presence of daytime hypercapnia, symptoms or signs (e.g., ALSFRS-R subscores, paradoxical breathing, orthopnea) indicating clinically relevant diaphragm weakness, or evidence of nocturnal hypoventilation. 86 COVID-19 has also influenced routine respiratory monitoring and NIV initiation, with less reliance on pulmonary function testing, as well as trends toward device titration in the outpatient or home setting. 87
Further details on NIV prescribing practices and patient utilization were evaluated longitudinally as part of a recent clinical trial of fast skeletal muscle troponin activators (VITALITY-ALS trial). Participating clinics were able to prescribe NIV without restriction to study participants, who at baseline had a vital capacity >70% predicted and did not use NIV. Upon enrollment, approximately one-third of participants were prescribed NIV for worsening vital capacity, sleep problems, or respiratory symptoms, and 70% of those prescribed tolerated >4 h NIV use per day. As vital capacity became severely affected (<50% predicted), nearly one-third of participants were not prescribed NIV. No differences in regional practices (USA, Europe) were identified. 88
Despite the evidence indicating NIV may preserve quality of life and prolong survival, ultimately the choice to initiate (or discontinue) external ventilation resides with patients, in conjunction their family and caretakers. Studies of the benefits and care burdens of NIV are limited but suggest perceived threats to autonomy, anxiety over NIV use and disease progression, and difficulties in navigating the healthcare system may steer patients away from NIV. In contrast, greater coping skills and resilience of caregivers may foster use. 89 Further work is also needed to determine whether technical difficulties (i.e., sialorrhea or the inability to adjust the mask with progressive motor involvement) contribute to lower utilization or refusal of NIV by patients.
Future studies
While cardiovascular and respiratory comorbidities of sleep dysfunction have been well documented, sleep dysfunction also appears to impact cognitive and non-respiratory motor function in other neurodegenerative conditions. For example, in Parkinson’s disease, longitudinal polysomnography studies identified deficits in short wave sleep that corresponded to cognitive decline. 90 Correlations between OSA and cognitive decline also exist for the general population and in other neurodegenerative disorders, such as Alzheimer’s disease. 91 Interestingly, a recent report found 3 of 10 “hub genes” that closely linked Alzheimer’s disease with sleep disorders have also been associated with ALS. For example, SOD1 was reported as a sleep deprivation and sleep disorders related gene. 92 A small clinical trial indicated 6 weeks of NIV corrected some cognitive dysfunction in patients with ALS. 93 Further study of neuroprotective benefits of early NIV on cognitive health and motor function in ALS are warranted.
Compared to previous rates of early ALS progression, recent COVID-19 related quarantines led to steeper, earlier declines in function (as measured by ALSFRS-R), along with rapid requirement for NIV and increased mortality. 94 The authors identified multiple contributors to faster disease progression, including delays in accessing resources such as NIV. Other contributors were limited access to in-person medical and rehabilitation care, greater social isolation, and reduced clinical trial enrollments. 94 As COVID-19 becomes endemic, it will be important to understand whether a restored access to care and resolution of supply chain problems stabilize ALS progression.
Respiratory strength training (RST)
Respiratory weakness and hypoventilation are consistent features of progressive neuromuscular conditions, such as ALS. Thus, there has been great interest in determining whether targeted exercises can improve respiratory function. Evidence-based respiratory training modes in neuromuscular disease include the use of pressure-threshold or electrically braked flow-resistive devices, as shown in Figure 4.95,96 Training intensity is typically prescribed by a physiotherapist or speech language pathologist, as a percentage of maximum inspiratory or expiratory pressure. These protocols are consistent with training specificity principles, in which short bouts of moderate to high-intensity exercises (typically an intensity of >50% of maximal respiratory pressures) are expected to recruit fast diaphragm motor units along with accessory respiratory motor neuron pools.97,98 Clinical studies of respiratory strength training indicate potential of RST to preserve or improve ventilation, cough, and swallowing function (Table 1).
99
Respiratory strength training is typically accomplished using portable devices that impose an external load to the muscles of inspiration or expiration. Right image: pressure-threshold devices typically use a calibrated spring and valve that blocks either inspiration or expiration. To open the valve, the patient must exert an increased effort to generate enough pressure to overcome the tension exerted by the spring. Left image: respiratory loads can also be delivered via flow resistance. Uncontrolled resistive loads, such as straw or impeded snorkel-breathing, can be minimized with a slow breath pattern with low airflows. To overcome this issue, some devices incorporate electronic braking algorithms capable of delivering a consistent, proportional load throughout the breath volume.
Diaphragm slow and fast fiber atrophy in ALS has been reported even with mildly reduced respiratory capacity. 100 Therefore, inspiratory strength training has traditionally been of considerable interest. Inspiratory strength training has been well-tolerated by patients and preserved or improved maximal inspiratory pressure (MIP) outcomes.101,102 However, unresisted diaphragmatic breathing alone does not appear to preserve inspiratory function. 103
An early clinical trial of inspiratory strength training compared 12 weeks of exercise to a usual care control, followed by 8 weeks of detraining. The training group used completed 3, 10-min exercise sessions that gradually increased from 15% to 60% of sniff nasal inspiratory pressure. There were no serious adverse effects of training, and compliance was high. 104 Training yielded a modest, non-significant increase in MIP that dissipated following the detraining period.
Pinto and colleagues 105 investigated short- 105 and longer-term 106 effects of inspiratory strengthening exercises, in patients with early-stage ALS. The active strengthening group performed 2, 10-min exercise sessions daily for 8 months, at an intensity 30–40% of MIP. Another group trained at the lowest training intensity (9 cm H2O) for the first 4 months, followed by 4 months of IMT. No short-term between-group differences were found in ALS-FRS, the primary outcome, or in other maximal respiratory tests. In a follow-up study, a survival analysis compared those who continued IMT (up to 32 months) to a historical control. Significant independent predictors of mortality included absence of IMT, lower baseline phrenic response to magnetic stimulation, and male gender. 106
Respiratory strengthening has also been investigated to preserve airway protection behaviors. Plowman and co-workers hypothesized that expiratory strengthening would significantly improve maximal expiratory pressure (MEP), as well as swallowing and cough function. In this randomized, sham-controlled trial, 48 patients were allocated to either 8 weeks of expiratory strengthening or sham training with a deactivated device. Those who completed expiratory strength training significantly improved MEP and exhibited less pharyngeal residue during modified barium swallow testing. However, group differences were not observed in vital capacity or cough force. 107
Recent developments
A recent pilot clinical trial analyzed 8 weeks of remotely supervised inspiratory muscle training by the POWERbreathe®, an electronically-braked inspiratory resistive device. Training included 15 deep breaths, twice-daily; intensity gradually increased from 30% to 60% of MIP and was compared to a group that received usual care. Training led to a significant increase in MIP in the experimental group, accompanied by a reduced resting heart rateand improved heart rate variability. 108 A low heart rate variability reflects altered sympathovagal balance, which appears disturbed in those with ALS with severe pulmonary involvement. 109
Future studies
The durability of IMT on heart rate variability and autonomic function are poorly understood; further study is needed to assess its clinical significance. While cough force has traditionally been reinforced with expiratory strengthening, inspiratory flow and pressure generation are also significant predictors of cough flow in ALS. 110 Further investigation is indicated to identify whether combined inspiratory and expiratory training, as opposed to either mode in isolation, could more effectively enhance ventilation and airway protection function in ALS. In addition to research on combined training, training-induced changes in patient-reported outcomes have not been examined in ALS. In pulmonary conditions such as emphysema, RST has been associated with reduced diaphragm activation during exercise, 111 improved dyspnea 112 and dyspnea-related fear of movement. 113 It is not known if training reduces dyspnea in ALS. While one criticism of RST is the primary outcome measures are typically effort- and learning-dependent, a non-voluntary measure of diaphragm/phrenic function, such as diaphragm ultrasound or diaphragmatic pressure responses to twitch stimulation, could delineate effort-independent changes in function and help distinguish preservation of phrenic versus accessory motor neuron pools.
Therapeutic acute intermittent hypoxia (tAIH)
In contrast to the direct effects of RST on modulating respiratory neuromuscular output, therapeutic acute intermittent hypoxia (tAIH) is an approach aimed at eliciting synaptic plasticity and long-term facilitation (LTF) of breathing, which is a facilitation that emerges and persists after the therapeutic stimulus has ended. tAIH signaling pathways have been well-characterized in the phrenic motor system of animal models.114–117 As depicted in Figure 5, tAIH consists of mild, brief fluctuations in the concentration of inspired oxygen (usually 10–12% O2 for 1–2 min), interspersed with normal oxygen breathing (∼21% O2 for 1–2 min). Hypoxia-induced carotid body activity excites brainstem respiratory nuclei that transiently increase minute ventilation. In addition, tAIH simultaneously induces serotonin release, via the raphe nuclei, which then initiates cell signaling cascades that trigger brain-derived neurotrophic factor synthesis. The brain-derived neurotrophic factor synthesis is necessary for synaptic plasticity.114,116 With respect to the phrenic motor system, synaptic plasticity and LTF of breathing are expressed as gains in phrenic neural output, resting breathing, and maximal chemically stimulated breathing (Table 1).114,115 Moreover, tAIH also appears to promote neuroplasticity of non-respiratory motor pools. AIH protocols consist of several (typically 5–15) short intervals of breathing mildly lowered oxygen, interspersed with breathing normal air. Hypoxia durations used in patient populations vary from 1–4 min in duration and are typically between 9–12% oxygen, which is equivalent to altitudes found at Pike’s Peak (∼12.3% O2) to Mount Denali (∼9.7% O2). Periods of normoxia (21% O2) may differ between patient populations, but typically last between 1-4 min. Researchers have used custom-mixed gas bags or commercially-available altitude simulators to deliver lowered oxygen air at the desired concentration. The effects of single AIH sessions may emerge 30–60 min after the last hypoxic interval and may last for several hours.
The dose of AIH is critical in determining the direction and extent of plasticity, as well as the potential for undesired effects 118 Severe and prolonged intermittent hypoxia operates through different signaling pathways to simulate effects of obstructive sleep apnea and can lead to systemic hypertension, neuroinflammation, and cognitive impairment. 119 In contrast, serotonin-mediated low dose tAIH elicits neuroplasticity without detectable pathology,120,121 making it a potential candidate for promoting rehabilitation from neurological injury or disease. Further, tAIH is simple to apply and well tolerated by most individuals. 122
The effects of tAIH on human respiratory function were first investigated by Tester et al. in people with chronic spinal cord injury (SCI), subjects were exposed to 8, 2-min intervals of 8% inspired O2 for 10 consecutive days. Significant and sustained increases in minute ventilation persisted following each tAIH exposure. Sham hypoxia exposures did not yield similar increases in ventilation 123 and provided proof-of-principle that tAIH regimens could be used to enhance breathing function in adults with SCI. 124 Further evidence in patients with SCI indicates tAIH also induces non-respiratory motor plasticity, leading to gains in upper and lower extremity strength and walking function.124–129 Single tAIH sessions trigger acute, transient increases in hand or ankle for minutes to hours in duration.125,128 Other various studies indicate daily AIH, when combined with task-specific skilled rehabilitation, led to improved hand dexterity, gait speed, and walking endurance; these than daily AIH treatment alone.126,127 Larger walking gains occurred in those who needed more walking support at baseline. 129 As revealed by these studies, the intermittent release of serotonin and subsequent new synthesis of brain-derived neurotrophic factor, rather than the low oxygen, per se, are purported to facilitate these motor gains.130,131
Proof-of-concept studies of tAIH in ALS demonstrated that phrenic LTF was enhanced in end-stage SOD1G93 A rats. Interestingly, tAIH restored most of the rodents’ capacity to generate phrenic motor output.132–134 This evidence of tAIH-induced phrenic neural plasticity in ALS rodents, combined with the documented rehabilitation potential in chronic SCI suggested a similar potential to elicit breathing plasticity in patients with ALS.
Recent developments
In the first study in tAIH in people with ALS, Sajjadi and colleagues 135 examined tAIH effects on collective respiratory muscle activity and breathing. The authors demonstrated that, when compared to a session where subjects were administered sham tAIH, a single tAIH session enhanced tidal volume, minute ventilation and collective respiratory muscle activity during quiet breathing 1 h after the session was completed. Multi-channel surface EMG recordings were measured by a validated vector analysis approach, to calculate the cumulative magnitude and pattern of recruitment. 136 Breathing improvements were noted in ALS subjects as well as older adult controls, and participants did not report breathlessness or discomfort during hypoxic episodes. Although tAIH did not alter MIP and some between-subject variation was noted, the authors concluded that tAIH was safe, feasible, and it warrants further controlled study to determine if tAIH could prolong independent breathing in people living with ALS. A sub-investigation of the same study evaluated changes in inspiratory drive following tAIH, as measured by inspiratory occlusion pressure (P0.1). Although P0.1 remained consistent 1 h following tAIH, inspiratory pressure generation at 300 ms into inspiration was significantly greater in those with ALS. The authors theorized that, while underlying respiratory drive was not impacted by tAIH, those with ALS had a greater ability to detect and volitionally respond to an inspiratory occlusion post-tAIH. Further, the authors posited that compensatory responses to inspiratory occlusions may provide additional insights into the ability of patients to quickly generate pressure when challenged. 137 As indicated previously, the tAIH “dose” is critical for promoting serotonergic signaling, while avoiding negative effects of severe hypoxia. While some clinical trials have used commercially available altitude simulators to deliver tAIH, the intervention requires supervision for vital sign monitoring and patient tolerance. Moreover, the optimal tAIH protocols in humans remain under investigation in multiple research labs.
Future studies
Sajjadi and co-workers hypothesized that adenosine-mediated signaling could be responsible for the variability in ventilatory LTF of subjects, since adenosine signaling is often elevated in people with ALS 138 An ongoing clinical trial (NCT05377424) 139 will investigate whether pretreatment with istradefylline, a selective adenosine 2A receptor (A2AR) antagonist, can further amplify tAIH-induced breathing plasticity in those with ALS. This work is supported by studies in ALS rodent models 134 and patients with SCI 140 illustrating that pre-treatment with an A2A receptor inhibits A2AR-mediated cellular signaling during tAIH and restores serotonergic-mediated AIH neuroplasticity. 141 It is not known whether tAIH in ALS may facilitate function of motor neuron pools involved in airway protection or limb function. Finally, the feasibility and durability of repeated tAIH on longer-term preservation of breathing remains untested and requires further examination, prior to integration into clinical practice.
Conclusion
Degeneration of respiratory motor neurons leads to progressive dystussia, dysphagia, and hypoventilation in those with ALS. The respiratory therapies for ALS summarized in this review, including mechanical insufflation-exsufflation, lung volume replacement, noninvasive ventilation, and respiratory strength training can be applied in conjunction with pharmacological approaches, to help stabilize respiratory function in ALS. Current knowledge and recent research developments highlight the feasibility and safety of these non-pharmacological respiratory therapies. As part of patient-centered ALS care, regular monitoring of pulmonary function and respiratory strength, resting capnography and oxygen saturation, swallowing function, sleep, dyspnea and related patient reports can detect subtle changes in respiratory function and trigger initiation of suitable respiratory treatments. Members of the interprofessional respiratory care team include physicians (neurology, pulmonology, otolaryngology, sleep medicine), respiratory and physical therapists, speech language pathologists, dieticians, case managers, and medical equipment specialists. Therapeutic acute intermittent hypoxia is a recently reported non-pharmacological therapy that aims to promote neuroplasticity of breathing yet requires more extensive proof-of-principle testing in ALS. Future research can help elucidate the doses and optimal timing for initiating respiratory therapies and identify patients most likely to yield the greatest clinical benefit. By addressing these questions, future work can improve the quality of life and survival of those living with ALS.
Footnotes
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research,authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article: PSD was supported by funding from the ALS Association. WO was supported by T32 HL134621.