
Abstract
Keywords
Introduction
Diabetes mellitus (DM), as a chronic and globally prevalent metabolic disorder, is associated with an increased risk of cardiovascular morbidity and mortality. 1 According to the IDF Diabetes Atlas 2021, there were 537 million adults living with diabetes worldwide, and this number is predicted to rise to 783 million by 2045. In 2021 alone, diabetes was responsible for 6.7 million deaths. 2 Patients with DM continue to be at risk of vascular inflammation and injury even after achieving glycemic control. Advanced glycation end products (AGEs), derived from nonenzymatic glycation of proteins and lipids, are not easily degraded in vivo and can accumulate in diabetic tissues for extended periods. 3 AGEs can therefore explain the occurrence and progression of diabetic vascular complications resulting from prolonged exposure to high glucose levels. Recent studies have indicated that AGEs play a significant role in the development of vascular pathology. 4 However, the precise mechanisms underlying the relationship between AGEs and vascular pathology remain elusive.
The large conductance calcium-activated potassium (BK) channels rigorously regulate the membrane potential and tone of coronary arterial smooth muscle. 5 BK channels are composed of four α-subunits (BK-α), auxiliary β-subunits (BK-β), and/or γ-subunits (BK-γ). The BK-β subunit enhances channel sensitivity to Ca2+ and participates in the regulation of channel electrophysiology.6,7 It has four subtypes, with the β1 subtype being abundantly expressed in coronary arterial smooth muscle cells (SMCs). 8 Our previous study demonstrated that BK channel currents account for approximately 65% of the total potassium currents in rat coronary arteries. 9 Considerable evidence suggests that the downregulation of BK-β1 subunit in diabetes is primarily responsible for BK channel dysfunction.5,10 It has been reported that AGEs impair the expression and function of Kv channels in rat coronary SMCs, resulting in Kv-mediated coronary dysfunction in diabetic rats. 11 However, it remains unclear whether AGEs decrease the expression of BK-β1 subunit, and the underlying mechanism also needs to be investigated.
The purpose of our study was to investigate whether AGEs aggravated BK channel dysfunction and the underlying mechanisms. We found that AGEs could inhibit the phosphorylation of adenosine 5’-monophosphate-activated protein kinase (AMPK) and protein kinase B (PKB or Akt) in diabetic rats, which played an important role in the degradation of BK-β1 mediated by F-box only protein32 (FBXO32). This ultimately impaired BK channel function and coronary vasodilation. However, inhibition of AGEs generation by aminoguanidine upregulated the protein expression of BK-β1 and improved coronary function.
Methods
Experimental animal models
Sprague-Dawley rats weighing approximately 200 g (n = 80) were obtained from Changzhou Cavens Laboratory Animal Company in China. The rats were housed in pathogen-free conditions and had free access to standard food and water. The rat diabetic model was established as previously described. 12 Briefly, the rats received an intraperitoneal injection of streptozotocin (STZ; Sigma-Aldrich Corp.; 60 mg/kg) for two consecutive days. One week later, after fasting for 6 h, rats with blood glucose concentrations greater than 16.7 mmol/L were enrolled in this study. The diabetic rats were then randomly divided into two groups: DM + aminoguanidine (DM+A) group (100 mg/kg/d, treated by intragastric administration, n = 20), and DM+Vehicle (DM+V) group (treated by intragastric administration of 0.9% NaCl, n = 20). The control group, treated in the same manner, was also divided into two groups: Control + aminoguanidine (C+A) group (n = 20) and Control + Vehicle (C+V) group (n = 20). After 8 weeks, the rats were sacrificed, and their coronary arteries were harvested and stored at -80°C. The animal experiments were conducted according to the NIH Guide for the Care and Use of Laboratory Animals (NIH Publication 8th edition, Washington, DC: National Academies Press, US, 2011). The study protocol was approved by the Institutional Animal Care and Use Committee of Wuxi People’s Hospital affiliated with Nanjing Medical University.
AGEs concentration measurement in vivo
After a 24 h fasting period, blood samples were collected from the inferior vena cava of the rats to determine the levels of AGEs. The concentration of plasma AGEs was measured using a Rat AGEs enzyme-linked immunosorbent assay (ELISA) kit (Mlbio, #ML003305).
Isolation of rat coronary arteries
The rats were anesthetized with isoflurane (2%) (Ruiwode Lift Technology Co.) inhalation, and the hearts were rapidly removed and placed in an ice-cold physiological saline solution (PSS), which contained NaHCO3 14.9 mmol/L, NaCl 130 mmol/L, CaCl2 1.6 mmol/L, MgSO4•7H2O 1.17 mmol/L, KCl 4.7 mmol/L, KH2PO4 1.18 mmol/L, Glucose 5.5 mmol/L, EDTA 0.026 mmol/L (pH 7.4 adjusted with HCl). Then, the right and left coronary arteries, as well as the septal coronary arteries, were carefully dissected and freed from surrounding myocardium and connective tissue under a microscope (Olympus SZX10 Stereo Microscope). All the chemicals mentioned above were obtained from Sigma-Aldrich Corporation.
Measurements of coronary artery tension
For coronary artery tension measurement, the isolated coronary arteries were cut into arterial rings with a length of 1.5–2.0 mm. They were then threaded onto two tungsten wires and mounted in a four-chamber wire myograph (model 620M, Danish Myo Technology). The coronary arteries were maintained at 37°C in PSS. The PSS was pre-warmed to 37°C and gassed with 95% O2 and 5% CO2 to maintain a pH of 7.4, while a resting tension of 2.5 mN was applied, as previously reported. 12 The vessels were allowed to equilibrate for 60 min, with the solution being changed every 15 min. 13 Subsequently, the vessel tension was detected using a potassium-rich solution (60 mmol/L KCl) and recorded as the maximum contraction for contractile capacity. Once the vessels achieved a stable contractile response, the chambers were washed three times with PSS to restore basal tension. Then, a specific inhibitor of BK channels (iberiotoxin, IBTX) was added to the chambers. The changes in rat coronary tension were recorded using a setting of 100 nmol/L IBTX. All the chemicals mentioned above were obtained from Sigma-Aldrich Corporation.
Enzymatic isolation of rat coronary arterial SMCs
The isolated rat coronary arteries were dissected in ice-cold dissociation buffer containing 145 mmol/L NaCl, 4 mmol/L KCl, 1 mmol/L MgCl2, 0.05 mmol/L CaCl2, 10 mmol/L glucose, and 10 mmol/L HEPES (pH 7.4 adjusted with NaOH). Three dissected coronary arteries were placed in 1.0 mL of physiological saline solution containing bovine serum albumin (1 mg/mL). Then, they were transferred into another tube and incubated with 1.5 mg papain, 1.0 mg dithiothreitol, and 0.1% bovine serum albumin in 1.0 mL of saline solution. Subsequently, they were followed by incubation with 1.0 mg collagenase, 1.0 mg trypsin inhibitor, 0.25 mg elastase, and 0.1% bovine serum albumin in 1.0 mL of saline solution. Each step was performed for 9 min at 37°C in a shaking water bath. The coronary arteries were then washed three times with saline solution, placed in saline solution at 4°C, and used on the same day. The SMCs of the coronary arteries were obtained before performing the experiments by gently pipetting with a transferpettor until they were completely dissociated. All the chemicals mentioned above were purchased from Sigma-Aldrich Corporation.
Whole-cell BK channel current recordings
Whole-cell BK currents were recorded from freshly isolated rat coronary arterial SMCs using an Axopatch 200B Amplifier (Molecular Devices, Inc.). The recordings were filtered at 2 kHz and sampled at 50 kHz, as previously reported. 9 All experiments were conducted at room temperature (22–24°C). Whole-cell BK currents were defined as the IBTX (100 nmol/L)-sensitive K+ current component by digital subtraction. Whole-cell K+ currents were elicited from a holding potential of −60 mV with 100 ms duration pulses to testing potentials ranging from −40 mV to +160 mV in 10-mV increments. After obtaining normal rat coronary arterial SMCs, some cells were directly isolated for BK current density detection, while others were incubated with AGEs for 2 h. These groups were defined as the Ctrl group and AGEs group, respectively. The BK channel current density was recorded for all four rat groups. For whole-cell BK channel recordings, the extracellular solution contained 145.0 mmol/L NaCl, 5.6 mmol/L KCl, 1.0 mmol/L MgCl2, 0.5 mmol/L CaCl2, 10.0 mmol/L HEPES, and 10.0 mmol/L glucose, at pH 7.4. The pipette solution contained 140.0 mmol/L KCl, 0.5 mmol/L MgCl2, 1.0 mmol/L Na2ATP (EGTA), and 0.465 mmol/L CaCl2 (∼200 nmol/L free Ca2+), at pH 7.35. All chemicals mentioned above were obtained from Sigma-Aldrich Corporation.
Cell culture protocol
Human coronary arterial SMCs (ATCC, #PCS-100-021) and the culture medium (ATCC, #PCS-100-042 and #PCS-100-030) were purchased from ATCC. The cells were seeded into culture plates (Corning, #430167) and initially incubated at 37°C with 5% CO2. When the cells reached 90% confluence, they were subcultured onto new culture dishes. The culture medium was changed every 2−3 days. To investigate the effect of advanced glycation end products (AGEs) on BK channel expression, human coronary arterial SMCs were exposed to 100 μg/mL AGEs-bovine serum albumin (AGEs-BSA) (Abcam, #ab51995) for 48 h. Cells cultured with 100 μg/mL non-glycated BSA were used as a BSA control. 11 To further explore the mechanisms by which AGEs regulate BK protein expression, the cells were divided into four groups: the normal glucose group (NG), the high glucose group (HG), the NG group cultured with aminoguanidine (NG+A), and the HG group cultured with aminoguanidine (HG+A). Cells in the NG group were exposed to 5.5 mmol/L glucose, and cells in the HG group were exposed to 25.5 mmol/L glucose. Aminoguanidine at 100 μmol/L was used as an AGEs crosslink breaker. 14 The cells were incubated for 96 h in 6-well plates for the western blotting assays and quantitative real-time (qRT) PCR. To investigate the mechanisms involved in the AMPK/Akt signaling pathway in human coronary SMCs, Compound C (Abcam, #ab120843), an inhibitor of AMPK, and MK2206 (Beyotime, #SF2712), an inhibitor of Akt, were used to modulate the pathway activity.
Quantitative real-time (qRT) PCR
Total RNA was extracted from rat aortic arteries and four groups of cells. The RNA was then reverse-transcribed using a reverse transcription system (Takara Bio. Inc., #DRR036A). Quantitative PCR was performed on an ABI Prism 7500HT sequence detection system (Applied Biosystems Fisher Scientific Inc.) using a SYBR Green PCR mix (Roche Co., #4913914001). The cycling conditions were set as follows: step 1: 50°C for 2 min; step 2: 95°C for 10 min; and step 3: 95°C for 15 s, followed by 60°C for 60 s, repeated for 40 cycles. β-actin was used as an internal control to normalize the differences in total RNA amount among the rat samples, while GAPDH was used as the internal control in each group of cells. The relative mRNA expression levels of the target genes were calculated using the 2−ΔΔCt method. The primer pairs used for cDNA amplification were as follows: Rat BK-β1 forward: 5’-CCAACAGTGCTCCTATATCCCCA-3’, reverse: 5’-ATAAGAAGGCCACCAGTCAGCAG-3’; Rat BK-α forward: 5’-AAACAAGTAATTCCTCAAGCTGGTG-3’, reverse: 5’-CGTAAGTGCCTGGTTGTTTTGG-3’; Rat β-actin forward: 5’-AGATTACTGCCCTGGCTCCTA-3’, reverse: 5-CCTGCTTGCTGATCCACATCT-3’; Human BK-β1 forward: 5’-GACCAACATCAGGGACCAGG-3’, reverse: 5’-GGCCGTCTGGTAATTGTCCA-3’; Human BK-α forward: 5’-GGGTGATGATATCCGCCCAG-3’, reverse: 5’-AAACCGCAAGCCGAAGTAGA-3’; Human GAPDH forward: 5’-GTCAAGGCTGAGAACGGGAA-3’, reverse: 5’-AAATGAGCCCAGCCTTCTC-3’.
Western blot analysis
The isolated rat coronary arterial segments and cultured cells were lysed in ice-cold RIPA buffer (Pierce Co., #89900) containing protease and phosphatase inhibitors (Roche Co., #04693159001). The lysates were subjected to electrophoresis using SDS-PAGE and transferred onto polyvinylidene difluoride (PVDF) membranes (Roche Co.). The PVDF membranes were then incubated with specific primary antibodies, including GAPDH (Cell Signaling Technology Co., #2118), BK-α (Santa Cruz Biotechnology, #sc-374,142), BK-β1 (Alomone Labs, #APC-036), anti-AMPK antibody (Cell Signaling Technology, #2532), anti-p-AMPK antibody (Cell Signaling Technology, #2535), anti-FBXO32 antibody (Abcam, #ab194791), anti-Akt antibody (Cell Signaling Technology, #4691), and anti-p-Akt antibody (Cell Signaling Technology, #4060). Immunoblot bands were quantified using densitometry analysis with ImageJ software (Scion Corp.). The densities were normalized to the levels of the control treatment, and the relative folds were normalized to GAPDH levels.
Statistical analysis
SPSS 22.0 (Statistical Product and Service Solutions 22.0, IBM) and Graphpad Prism 7.00 (GraphPad Software, Inc) were used for statistical analysis. Data were presented as mean ± SEM (standard error of mean). Student's t-test was used to compare data between two groups. One-way ANOVA with post-hoc LSD analysis was used to compare data between multiple groups. p value <.05 was considered statistically significant.
Results
AGEs aggravated rat coronary arterial dysfunction
Basic data of the four rat groups after modeling.

n = 20 for each group. Data are presented as mean ± SEM.
ap < .05 versus C+V.
bp < .05 versus C+A.
cp < .05 versus DM+V.

Effects of inhibition of AGEs on coronary artery tensions and BK channel densities and protein expression (a) Representative tracings for 60 mmol/L KCl and 100 nmol/L IBTX induced vascular tension alterations of coronary arterial rings from C+V, DM+V, C+A and DM+A groups. (b) Graph data showing the vascular tension alterations induced by KCl. (c) Graph data showing the vascular tension alterations (IBTX/KCl). (d and e) Whole-cell potassium currents before and after application of 100 nmol/L IBTX, and the I-V relationship of IBTX-sensitive currents of control and AGEs-cultured freshly isolated rat coronary arterial SMCs (n = 3∼6 per group). (f) The representative tracings of baseline potassium currents and potassium currents after application of 100 nM IBTX in rat coronary arterial SMCs of the C+V, DM+V, C+A and DM+A groups, respectively (n = 3∼5 per group). (g) Graph data showing IBTX-sensitive current densities at the testing potential of +100 mV in rat coronary arterial SMCs of the four groups. (h–j) The protein expressions of BK-α and BK-β1 in human coronary arterial SMCs in the BSA and BSA-AGEs groups (n = 6∼9 per group). Quantitative analysis of BK-α and BK-β1 were normalized to GAPDH protein expression levels. (k-l) The mRNA expression of BK-α and BK-β1 in rat coronary arteries of the C+V, DM+V, C+A and DM+A groups. β-actin was used as an internal control to normalize differences in the amount of total RNA in each rat sample (n = 4 per group). (m and n) The mRNA expression of BK-α and BK-β1 in human coronary arterial SMCs of the NG, HG, NG+A, HG+A groups. GAPDH was used as an internal control to normalize differences in the amount of total RNA in each cell sample (n = 4∼5 per group). (o–q) Protein expressions of BK-α and BK-β1 in rat coronary arteries of the C+V, DM+V, C+A and DM+A groups. Quantitative analysis of BK-α and BK-β1 were normalized to GAPDH protein expression levels (n = 5 per group). (r–t) Protein expressions of BK-α and BK-β1 in human coronary arterial SMCs of the NG, HG, NG+A, HG+A groups. Quantitative analysis of BK-α and BK-β1 were normalized to GAPDH protein expression levels (n = 5∼9 per group).
Adverse effects of AGEs on coronary arterial BK channel activation
To investigate the harmful effects of AGEs on BK channel function, whole-cell BK currents were recorded using the patch clamp technique. Figure 1(d and e) show the total K+ currents in rat coronary arterial SMCs from the Ctrl and AGEs groups at baseline and after exposure to 100 nmol/L IBTX. There was a decreasing trend in total K+ current densities in the AGEs group compared to the Ctrl group, but no significant difference was observed between the two groups. However, there was a significant reduction in BK current densities in the AGEs group, from 54.27 ± 14.83 pA/pF at a testing potential of +100 mV (p < .05, n = 3) in the Ctrl group to 16.31 ± 5.47 pA/pF (p < .05, n = 5) in the AGEs group. Figure 1(f and g) show the total K+ currents in rat coronary arterial SMCs from the four rat groups at baseline and after exposure to 100 nmol/L IBTX. There was a decreasing trend in total K+ current densities in the DM+V and DM+A groups compared to the C+V and C+A groups, but no significant differences were observed among the four groups. The current densities of BK channels decreased in the DM+V group (9.71 ± 2.58 pA/pF) compared to the C+V group (34.05 ± 5.46 pA/pF), while the decrease was significantly reversed in the DM+A group (30.19 ± 2.81 pA/pF). The current-voltage relationships confirmed that AGEs significantly reduced the BK channel current density. BK channel function was impaired in the DM+V group compared to the C+V group, and aminoguanidine could improve it by inhibiting the generation of AGEs.
AGEs promoted the down-regulation of BK-β1 subunit
Figure 1(h–j) show a decrease in BK-β1 protein expression in human coronary arterial SMCs detected through western blotting in the AGEs-BSA group compared to the BSA group. There was no significant difference in the protein expression of BK-α between these two cell groups. In Figure 1(k–n), the mRNA level of BK-β1 significantly decreased in DM+V rats, but increased in the DM+A group. However, there were no significant differences in the mRNA level of BK-α among the four rat groups. Additionally, there were no significant differences in the mRNA expression of BK-α and BK-β1 among the four human coronary arterial SMCs groups. In Figure 1(o–q), BK-β1 protein expression significantly decreased in the DM+V group compared to the C+V group. However, when the rats were treated with aminoguanidine, the BK-β1 protein expression was higher in the DM+A group than in the DM+V group. There were also no significant differences in BK-α protein expression among the four rat groups. The protein expression of BK-α and BK-β1 in the four human coronary arterial SMCs groups was consistent with that observed in vivo (Figure 1(r–t)). These data indicate that AGEs downregulate BK-β1 protein expression, while inhibiting AGEs generation can reverse and upregulate its expression. AGEs impair coronary BK channel function by down-regulating BK-β1 expression.
Effects of AGEs on FBXO32
Previous studies have demonstrated that the ubiquitination and degradation of BK-β1 subunit by FBXO32 can result in a decrease in BK-β1 expression. To investigate the effects of AGEs on FBXO32, we examined FBXO32 protein expression. The BSA-AGEs group showed increased FBXO32 protein expression compared to the BSA group (Figure S1(a and b)). In Figure 2(a–d), it is shown that the expression of FBXO32 was up-regulated in the DM+V group and down-regulated in the DM+A group after the application of aminoguanidine in vivo. The FBXO32 expression in vitro was consistent with that observed in vivo. Regulation of Akt in AGEs-mediated FBXO32-induced BK-β1 degradation (a and b) Protein expression of FBXO32 in rat coronary arteries of four groups (n = 5 per group). (c and d) Protein expression of FBXO32 in human coronary arterial SMCs of four cell groups. Quantitative analysis of FBXO32 was normalized to GAPDH protein expression levels. (e–g) Phosphorylation levels of Akt and total Akt in rat coronary arteries of four groups (n = 8 per group). (h–j) Phosphorylation levels of Akt and total Akt in human coronary arterial SMCs of four groups (n = 3 per group). The phosphorylation level of Akt (k and n) and the protein expressions of FBXO32 (l and o) and BK-β1 (m and p) were measured after human coronary arterial SMCs were incubated for 96 h in DMEM containing 25.5 mmol/L glucose, or 25.5 mmol/L glucose with aminoguanidine in the absence or presence of MK2206 (0.3 μM) (n = 5∼10 per group). MK2206 was added at the beginning and remained for 6 h
Regulation of AGEs-induced BK-β1 and FBXO32 expression by Akt
To investigate the role of AGEs in inhibiting Akt activation, we examined the expression of phosphorylated Akt and total Akt in human coronary arterial SMCs cultured with AGEs. In comparison to the BSA group, the BSA-AGEs group exhibited a decrease in phosphorylated Akt expression. However, there was no difference in total Akt expression between the two groups (Figure S1(c–e)). In Figure 2(e–j), it is shown that the expression of phosphorylated Akt was down-regulated in the DM+V group and HG group. On the other hand, Akt was activated in both the DM+A group and HG+A group after the application of aminoguanidine.
In order to further investigate the role of Akt in AGEs-induced degradation of BK-β1, we utilized the Akt inhibitor MK2206 (0.3 μmol/L). We observed that when the HG+A group of human coronary arterial SMCs were treated with MK2206, the protein expression of phosphorylated Akt and the ratio of pAkt/Akt decreased. Additionally, FBXO32 expression increased, and the protein expression of BK-β1 decreased compared to the HG+A group (Figure 2(k–p)). These findings suggest that AGEs downregulate the protein expression of BK-β1 through the Akt/FBXO32 pathway.
Regulation of AMPK by AGEs in vivo and vitro
Numerous studies have demonstrated the crucial role of AMPK in vascular homeostasis. We investigated the activation of AMPK both in vivo and in vitro. In Figure S1 (f–h), it can be observed that the expression of phosphorylated AMPK (p-AMPK) significantly decreased in human coronary arterial SMCs exposed to AGEs. However, there was no significant difference in AMPK expression between the BSA and BSA-AGEs groups. As shown in Figure 3(a–f), AMPK activation was reduced in the DM+V group and HG group. However, after intervention with aminoguanidine, the phosphorylation of AMPK was up-regulated in the DM+A group and HG+A group. These findings indicate that AGEs inhibit AMPK activation in rat coronary arteries and human coronary arterial SMCs. Furthermore, reducing the generation of AGEs can promote AMPK activation.
Regulation of AGEs-induced BK-β1, FBXO32 and Akt expression by AMPK
In order to further investigate the effect of AMPK on AGEs-induced BK channel dysfunction, we utilized Compound C (CC, 1 μmol/L), a phosphorylated AMPK inhibitor. We divided the cells into three groups: HG group, HG+A group, and HG+A+CC group. As depicted in Figure 3(g-m), the administration of CC led to a decrease in phosphorylated AMPK expression, as well as the protein expressions of phosphorylated Akt and the ratio of p-Akt/Akt. Moreover, we observed an increase in FBXO32 expression in the HG+A+CC group. Additionally, the protein expression of BK-β1 decreased in the HG+A+CC group. These findings suggest that inhibiting AGEs can activate AMPK, and the inactivation of AMPK leads to a decrease in downstream p-AKT expression. Consequently, this upregulates FBXO32 expression, promoting the degradation of the BK-β1 subunit. Regulation of AMPK in Akt-mediated FBXO32-induced BK-β1 degradation by AGEs (a–c) Protein expression of p-AMPK and AMPK in rat coronary arteries from the four groups (n = 8 per group). (d–f) Protein expression of p-AMPK and AMPK in human coronary arterial SMCs from the four groups (n = 9 per group). Quantitative analysis of p-AMPK and AMPK was normalized to GAPDH protein expression levels. (g) Human coronary arterial SMCs were incubated for 96 h in DMEM containing 25.5 mmol/L glucose, or 25.5 mmol/L glucose and aminoguanidine in the absence or presence of Compound C (CC, 1 μM). Subsequently, the phosphorylation level of AMPK (h and i), AKT (j and k), and the protein expressions of FBXO32 (l) and BK-β1 (m) were measured (n = 8 and 9 per group). Quantitative analysis of FBXO32 and BK-β1 was normalized to GAPDH protein expression levels.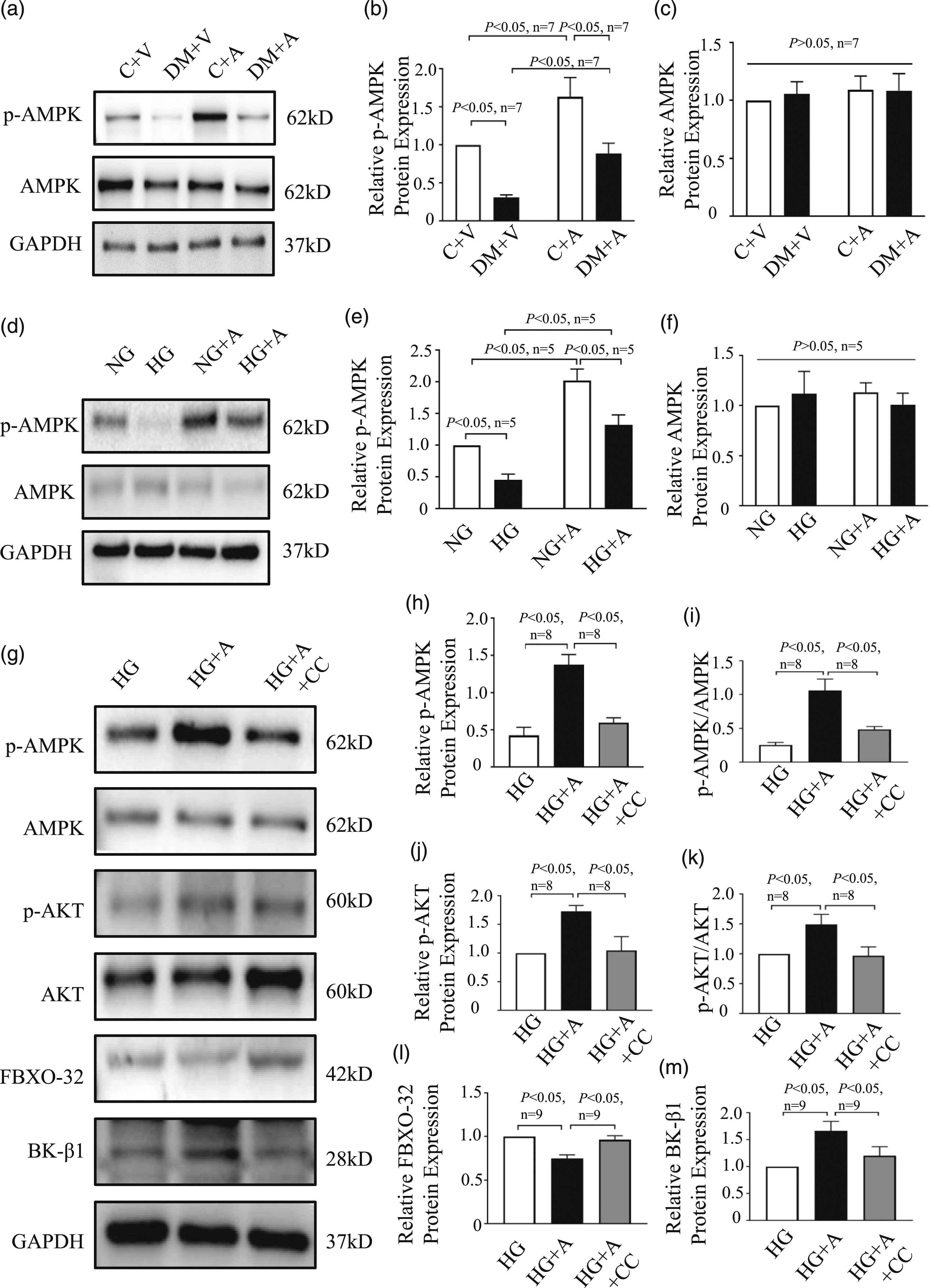
Discussion
Main findings
Our study reveals several key findings. Firstly, the results demonstrate that AGEs downregulate the protein expression of BK-β1, decrease BK channel currents, and ultimately impair coronary vasodilation function. Breaking or inhibiting AGEs formation can improve BK-mediated vasodilation in coronary arteries. Secondly, AGEs reduce the phosphorylation of AMPK and Akt, and upregulate the protein expression of FBXO32 in diabetic rat coronary SMCs and cultured human coronary arterial SMCs. Thirdly, the inhibitory effect of AGEs on upregulating BK expression is counteracted by AMPK and Akt inhibitors. In summary, the degradation of BK-β1 induced by FBXO32, combined with the inactivation of AMPK/Akt due to AGEs, contributes to the dysfunction of coronary artery vasodilation in diabetic rats (see Figure S2).
AGEs aggravate the impairment of BK channels due to FBXO32-induced BK-β1 degradation
The sustained accumulation of AGEs contributes to the phenomenon of metabolic memory observed in long-term follow-up clinical studies of diabetic patients. 15 Circulating levels of AGEs are associated with the morbidity and mortality of cardiovascular disease in both type 1 and type 2 diabetic patients.16,17 BK channels play a crucial role in regulating the membrane potentials and smooth muscle tone of coronary arterial SMCs.18,19 Extensive research has shown that vascular BK channel function is impaired, accompanied by a decrease in BK-β1 expression in diabetes.12,13 In this study, the serum concentration of AGEs was significantly increased in diabetic rats, and aminoguanidine was able to reduce AGEs concentration. We also observed a significant impairment in coronary artery function, along with a decrease in BK channel current density, in diabetic rats. After administering aminoguanidine via gavage, both coronary artery function and BK channel current density improved in the DM+A group. Furthermore, we observed a decrease in BK-β1 protein expression in response to AGEs both in vivo and in vitro. These findings suggest that the reduction in BK-β1 protein expression is responsible for the decrease in BK channel current densities, ultimately leading to coronary artery dysfunction in diabetic rats. Moreover, inhibiting AGEs generation can improve the coronary artery function of diabetic rats by upregulating the protein expression of BK-β1.
FBXOs are key components of the Skp1-Cullin-F-box (SCF) type ubiquitin ligase complex and serve as sites for enzyme-substrate interaction. 20 FBXO32 specifically binds to the PDZ-binding motif present in substrates, including most BK-β1 isoforms in various species, including humans. Previous studies have reported that FBXO32 contributes to the degradation of BK-β1 protein through the ubiquitin-proteasome system (UPS) in diabetic vessels.20,21 In our study, we observed a decrease in BK-β1 protein expression along with an increase in FBXO32 protein expression under AGEs or HG cultured conditions and in diabetic rats. The mRNA level of BK-β1 was found to be decreased in the DM+V group compared to the C+V group, while it increased in diabetic rats treated with aminoguanidine via gavage. However, exposure to different glucose concentrations, with or without aminoguanidine, did not alter the mRNA level. These results suggest that there may be an underlying mechanism through which AGEs decrease the expression of BK-β1 by activating FBXO32 and promoting UPS-mediated degradation.
Akt plays an important role in AGEs-related FBXO32-induced decrease of BK-β1 protein expression
The expression of FBXO32 is regulated by the forkhead box O family transcription factor (FOXO-3a), whose transcriptional activity is influenced by its subcellular localization. Akt phosphorylates FOXO-3a, leading to its exclusion from the nucleus and subsequent loss of transcriptional function. In conditions of DM or HG, the activity of Akt is diminished, 22 consequently enhancing FBXO32 expression and promoting the degradation of BK-β1. 21
In the cardiovascular system, Akt can be activated by various factors such as insulin, platelet-derived growth factor, and vascular endothelial growth factor. 23 It has been established that activated Akt can inhibit apoptosis in vascular s SMCs, indicating its protective effect on blood vessels. 24 Our findings revealed that in human coronary arterial SMCs cultured with AGEs, the phosphorylation level of Akt was significantly reduced, accompanied by increased FBXO32 expression. Similar trends in protein expression of FBXO32 and phosphorylated Akt were observed in streptozotocin (STZ)-induced diabetic rats and in SMCs cultured with HG, consistent with a previous study. 9 Following intervention with aminoguanidine, the phosphorylation level of Akt was significantly increased in diabetic rats and under HG culture conditions, leading to decreased FBXO32 expression. To investigate the role of Akt in AGEs-mediated reduction of BK-β1 expression, we utilized MK2206, an Akt inhibitor. MK2206 decreased Akt phosphorylation and increased FBXO32 expression in SMCs cultured with HG+AGEs+MK2206. Our results demonstrated that aminoguanidine activated Akt phosphorylation, decreased FBXO32 expression, and upregulated BK-β1 expression. However, inhibiting Akt activation reversed this process, indicating the involvement of Akt in the regulation of AGEs-mediated FBXO32-induced BK-β1 degradation.
AGEs aggravate Akt related FBXO32-induced BK-β1 degradation by inactivating AMPK
AMPK is a heterotrimeric protein kinase consisting of an α subunit encoded by PRKAA, a β subunit encoded by PRKAB, and a γ subunit encoded by PRKAG. The α subunit, which has two isoforms, serves as the catalytic subunit, while the β subunit (two isoforms) and γ subunit (three isoforms) act as regulatory subunits. These subunits can combine in 12 different ways. As a sensor of cellular energy changes, AMPK can be activated under conditions of energy deficiency and inhibited when there is an energy excess. Antidiabetic drugs such as metformin and SGLT2 inhibitors activate AMPK-dependent signaling pathways, thereby playing a protective role in cardiovascular diseases. 25 Furthermore, it has been reported that AGEs can inactivate AMPK.26,27 AMPK has been shown to function as an upstream kinase, phosphorylating Akt in cardiomyocytes, coronary artery SMCs, and coronary artery endothelial cells. 27 This, in turn, affects the expression of downstream proteins.
Exercise and low nutrition can also activate AMPK. High glucose levels can induce the dissociation of the AMPKα subunit from LKB1, resulting in AMPK dephosphorylation and subsequent inactivation. 28 Numerous studies have demonstrated the significant role of AMPK in vascular homeostasis. Schneider et al. 29 reported that AMPK could activate BK channels, leading to the hyperpolarization of the membrane in rat aortic SMCs. This, in turn, relaxes blood vessels and exerts an antihypertensive effect. Taguchi et al. 30 discovered that glucagon-like peptide-1 (GLP-1) significantly enhanced endothelium-dependent relaxation in diabetic aortas, potentially mediated by GLP-1 receptor-dependent activation of AMPK/Akt signaling pathway.
Our results demonstrated that inhibiting the generation of AGEs led to an increase in AMPK and Akt phosphorylation levels, a decrease in FBXO32 expression, and an enhancement of BK-β1 expression. To further investigate the role of AMPK in AGEs-induced dysfunction of BK channels, we added Compound C to the HG+A culture medium. Compound C was found to inhibit AMPK phosphorylation, subsequently suppressing Akt phosphorylation, increasing FBXO32 protein expression, and reducing BK-β1 expression. In summary, high glucose levels resulted in increased generation of AGEs, and our study suggested that AMPK is involved in the regulation of Akt-mediated FBXO32-induced degradation of BK-β1 by AGEs.
Study limitations
There are at least three limitations in this study. First, the animals in our study were STZ-induced T1DM rats. It is unknown whether the findings of this study can be extrapolated to patients with type 2 diabetes. Second, the mechanisms underlying the inhibitory effects of AGEs on AMPK phosphorylation need to be further explored. Third, the in vivo experiments suggest that DM can reduce the mRNA expression of BK-β1, but the in vitro experiments show that high glucose does not reduce the mRNA expression of BK-β1. This suggests that the reduction of BK-β1 expression in vivo may not solely be due to ubiquitination and degradation, but may also involve other factors affecting its transcription. These factors also require further study.
Clinical significance of the study
Our study has demonstrated that AGEs aggravate the dysfunction of coronary vasodilation by promoting BK channel dysregulation in T1DM rats. Therefore, inhibiting AGEs production is important in clinical treatment, in addition to intensive control of blood glucose in diabetic patients. Moreover, our study has shown that AGEs exacerbate BK channel dysfunction through the AMPK/Akt/FBXO32 signaling pathway. This suggests that targeting this signaling pathway may offer new potential therapeutic strategies in the future.
Conclusion
In conclusion, our study demonstrated that AGEs inhibit AMPK phosphorylation, resulting in decreased expression of phosphorylated Akt. This, in turn, upregulates FBXO32, leading to BK channel dysfunction. Based on these findings, AGEs may play a significant role as a risk factor in the development and progression of coronary diseases. Therefore, in addition to blood glucose control, inhibiting AGEs generation and targeting the signaling pathway may offer new potential therapeutic strategies for preventing and treating DM-related coronary diseases.
Supplemental Material
Supplemental Material - Advanced glycation end products impair coronary artery BK channels via AMPK/Akt/FBXO32 signaling pathway
Supplemental Material for Advanced glycation end products impair coronary artery BK channels via AMPK/Akt/FBXO32 signaling pathway by Xiao-Yan Li, Ling-Ling Qian, Ying Wu, Yu-Min Zhang, Shi-Peng Dang, Xiao-Yu Liu, Xu Tang, Cun-yu Lu and Ru-Xing Wang in Diabetes & Vascular Disease Research
Footnotes
Acknowledgements
The authors sincerely thank Dr. Tong Lu from Mayo Clinic in Rochester for his technical assistance.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by the National Natural Science Foundation of China (grant numbers: 81770331 and 82000317), Top Talent Support Program for Young and Middle-aged People of Wuxi Health Committee (No. BJ2020018), and Research Foundation from Wuxi Health Commission for the Youth (No. Q202034).
Author contributions
R-XW contributed to the experimental design. L-XY, Q-LL, W-Y, Z-YM, L-XY, D-SP, L-CY, and T-X conducted the experiments. L-XY and Q-LL analyzed the data. R-XW and L-XY drafted the manuscript. All authors reviewed and approved the final manuscript.
Supplemental material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.