
Abstract
Restenosis is a major limiting factor for a successful outcome in type 2 diabetes (T2D) patients undergoing percutaneous coronary intervention (PCI). The aim of this study is to explore the role and regulatory mechanism of FOS-like antigen 1 (FOSL1) in restenosis in T2D. A T2D with restenosis mouse model was established by the combination of high-fat diet and streptozotocin injection and by wire-injury. High glucose (HG)-treated vascular smooth muscle cells (VSMCs) were used to mimic T2D in vitro. The results of quantitative real time PCR and western blotting demonstrated that the expression of FOSL1 was increased not only in T2D mice or HG-induced VSMCs, but also in T2D mice that underwent wire-injury. HE staining revealed that FOSL1 knockdown significantly reduced the intimal/media ratio of T2D mice after wire-injury. Silencing of FOSL1 reversed the promoting effects of HG treatment on viability, migration and inflammation reactions, and the inhibiting effect on the apoptosis of VSMCs. Inhibition of ERK/AP-1 pathway obtained similar patterns in HG-induced VSMCs. The activation of ERK/AP-1 pathway reversed the influence of FOSL1 knockdown on HG-induced VSMCs. Our findings indicate that silencing of FOSL1 may suppress restenosis via regulation of the ERK/AP-1 pathway in T2D mice, pointing out a potential therapeutic target to prevent restenosis in T2D.
Introduction
Type 2 diabetes (T2D) is a major global public health concern with the increased risk of complications such as cardiovascular diseases (CVD). 1 Although therapies for CVD including percutaneous coronary interventions (PCI) have resulted in marked improvements, restenosis after vascular surgery remains a major challenge. 2 The abnormal proliferation and migration of vascular smooth muscle cells (VSMCs) and the concomitant inflammation are considered to exert an important role in restenosis progression. 3 Therefore, research on inhibition of the excessive hyperplasia of VSMCs and inflammation are crucial for improving the prognosis of T2D patients who have undergone PCI.
FOS-like antigen 1 (FOSL1) serves as an oncogene to affect cell proliferation and metastasis in several types of human cancers, such as lung, 4 prostate, 5 and breast cancers. 6 Meanwhile, FOSL1 also plays important role in angiogenesis. For instance, Weng et al. found enrichment of FOSL1 protein in hyperplastic tissues of patients with esophageal cancer restenosis. 7 Evellin et al. further demonstrated that overexpression of FOSL1 promotes angiogenesis in tumorigenesis via regulation of the integrins αv and β3. 8 In addition, Sobolev et al. 9 revealed that FOSL1 is upregulated in atherosclerosis (an important complication of T2D caused by VSMCs hyperplasia). However, the role of FOSL1 in atherosclerotic restenosis in T2D remains unknown.
It is acknowledged that ERK/AP-1 pathway is deeply involved in cellular processes of several malignant tumors.10–12 Kim et al. 10 reported that ERK/AP-1 is closely associated with MMP-9 mediated cell invasion in breast cancer. Fujisawa et al. constructed a mouse ovarian cancer model and uncovered that ERK/AP-1 regulated by IL-13 promotes the metastasis of cancer cells. 11 Zheng et al. 12 found that the activation of the ERK/AP-1 pathway remarkably accelerates cell proliferation in liver cancer. Additionally, FOSL1, also as an AP-1 transcription factor, is confirmed to interact with the ERK/AP-1 pathway to modulate proliferation and metastasis in osteosarcoma. 13 More importantly, Peng et al. 14 found that ERK/AP-1 can be activated by VPO1 to regulate VSMCs motility in human abdominal aortic aneurysm.
In the present study, the role of FOSL1 on restenosis in vitro and in T2D mice after wire-injury, and the interaction with ERK/AP-1 pathway were investigated. Our results reveal that silencing of FOSL1 represses the restenosis via the ERK/AP-1 pathway in TD2 mice and indicates an underlying target to prevent restenosis for T2D patients underwent PCI.
Materials and methods
Agents
Short hairpin (sh) RNA targeting FOSL1 (sh-FOSL1), sh-negative control (sh-NC), adenovirus vector (Ad)-sh-FOSL1, and Ad-sh-NC were purchased from Sangon Biotech (Shanghai, China). Streptozotocin (STZ) was obtained from Sigma Aldrich (San Luis, MO, USA). The RevertAid H Minus First Strand cDNA Synthesis kit, DyNAmo Flash SYBR Green qPCR kit, and apoptosis detection kit were all purchased from Thermo Fisher Scientific (Waltham, MA, USA). The primary antibodies (FOSL1, p-ERK, ERK, c-fos, c-jun, and GAPDH) and the HRP-conjugated secondary antibody used for western blotting were procured from Abcam (Cambridge, UK).
Animals
Male BALB/c mice (22–28 g, 20 weeks old) were purchased from EseBio, Co, Ltd (Shanghai, China) and allowed to adapt to the laboratory environment for 1 week. The experimental procedures were in strict accordance with the Guide for the Care and Use of Laboratory Animals and approved by the ethical committee of the Fourth Hospital of Hebei Medical University.
T2D mouse model establishment
Thereafter, the mice were divided into 2 groups (n = 10) ad libitum: the control group and the T2D group. The mice in the T2D group were initially fed with high-fat diet (3% cholesterol, 10% lard, 10% sugar, 10% egg yolk powder, and 67% basic diet) for 3 weeks. Subsequently, STZ (40 mg/kg/day) was intraperitoneally injected (i.p.) into the mice for consecutive 5 days. Afterwards, the level of fasting blood glucose was detected, which more than 16.7 mmol/L was determined as T2D mice. Meanwhile, the mice fed with normal diet were served as the control group.
Wire-injury for T2D mice
After construction of the T2D model, the mice were anaesthetized by pentobarbital sodium (50 mg/kg; i. p.). Wire-injury for common femoral artery was performed using a 0.36 mm guidewire as previously described 15 and then injected with Ad-sh-FOSL1 or Ad-sh-NC (1 × 1010 p.f.u.). The arteries of mice in the sham group underwent dissection, temporary clamping without passage of the wire. All the mice were sacrificed 14 days later. The common femoral artery tissues were collected and fixed by 4% paraformaldehyde or frozen directly in liquid nitrogen.
Hematoxylin-eosin staining
The fixed common femoral artery tissues were embedded in paraffin and sectioned at 4 μm thickness. The sections were deparaffinized with xylene, dehydrated with graded ethanol and stained with HE, and then observed by a light microscopy. The areas of intimal and media were measured using Zen 2009 image software, and the intimal/media ratio was calculated.
VSMCs isolation, culture and transfection
Primary VSMCs were isolated from the aortas of mice as the previous study mentioned. 16 The isolated VSMCs were cultured in Dulbecco’s Modified Eagle’s Medium (DMEM) containing 10% fetal bovine serum (FBS) and normal glucose (NG; 5.5 mmol/L) at 37°C with 5% CO2. To simulate T2D in vitro, VSMCs were cultured in DMEM containing 10% FBS and high glucose (HG; 25 mmol/L). The transfection experiments were performed using Lipofectamine 3000 (Invitrogen, Nanjing, China) based on the instructions. After transfection for 48 h, the cells were collected for the subsequent experiments.
Cell viability assay
The transfected HG-induced VSMCs were cultured in 96-well plates (2 × 103) for 24 h. Afterwards, approximately 20 μL MTT was added to incubate 2 h at 37°C. The absorbance values were detected by a micro-plate reader (Molecular Devices, Shanghai, China) at the wavelength of 450 nm.
Cell migration assay
For cell migration, the above VSMCs (1×105 cells/μl) were seeded in 12-well plates and grown approximately 100% confluence, followed by creating the wounds using a pipette tip. The cells were then incubated for 24 h at 37°C. The photomicrographs of the scratch wounds were captured using a light microscope and the relative migration rate was calculated.
Flow cytometric analysis
The apoptosis was analyzed by flow cytometry. In brief, the collected cells (1 × 105 cells/mL) were cultured in 96-well plates for 24 h, and then stained with V-FITC and PI using an apoptosis detection kit at 25°C for 20 min in the dark. The apoptotic cells were measured using a flow cytometer (BD Biosciences).
Quantitative real time PCR
Total RNA was extracted from the common femoral artery tissues of mice or HG-induced VSMCs using Trizol Reagent (Invitrogen). The cDNA was then synthesized using RevertAid H Minus First Strand cDNA Synthesis Kit, followed by performing qRT-PCR analysis with DyNAmo Flash SYBR Green qPCR Kit. The 2−ΔΔCt method was used to calculate the mRNA expression levels of FOSL1, MCP-1, TNF-α, and IL-6. GAPDH was served as the internal control.
Western blot assay
The common femoral artery tissues of mice or HG-induced VSMCs were lysed with RIPA buffer (Beyotime, Shanghai, China) and then determined the concentrations using Rapid Gold BCA Protein Assay Kit (Thermo Fisher Scientific), followed by separating the protein product using 10% SDS-PAGE and transferring onto PVDF membrane. The membrane was incubated with primary antibodies (1:1000) at 4°C overnight and then the secondary antibody for 1h at room temperature. GAPDH was used as the internal control. Immunoblotting was visualized using an ECL detection kit (Amersham Biosciences, Sweden).
Statistical analysis
Data were presented as means ± SD. SPSS 20.0 software (Chicago, USA) was used for statistical analysis. Student’s t-test and one-way/two-way ANOVA followed by Tukey’s multiple comparisons test were used for data analysis. P-value less than 0.05 indicated a statistically significant difference.
Results
Increased FOSL1 expression and inflammation responses are observed in T2D mice after wire-injury
The expression level of FOSL1 in femoral artery tissues of T2D mice and T2D mice after wire-injury was initially determined. We found that FOSL1 mRNA expression was increased in T2D mice compared to the controls (Figure 1(a), P < 0.05). Meanwhile, compared with the sham group, increased FOSL1 was also observed in T2D mice that underwent wire-injury (P < 0.05). Similar patterns were obtained in the protein level of FOSL1 measured by western blotting (Figure 1(b), P < 0.05). In addition, we also found relatively high levels of MCP-1, TNF-α, and IL-6 in femoral artery tissues of T2D mice and T2D mice after wire-injury (Figure 1(c), P < 0.05). Increased FOSL1 and inflammation responses are observed in T2D mice after wire-injury. (a) The expression of FOSL1 in T2D mice or T2D mice after wire-injury was detected by qRT-PCR. (b) The protein level of FOSL1 in T2D mice or T2D mice after wire-injury was measure by western blot assay. (c) The mRNA expression levels of MCP-1, TNF-α, and IL-6 in T2D mice or T2D mice after wire-injury were determined by qRT-PCR. *P < 0.05 vs. the Control group. #P < 0.05 vs. the Sham group.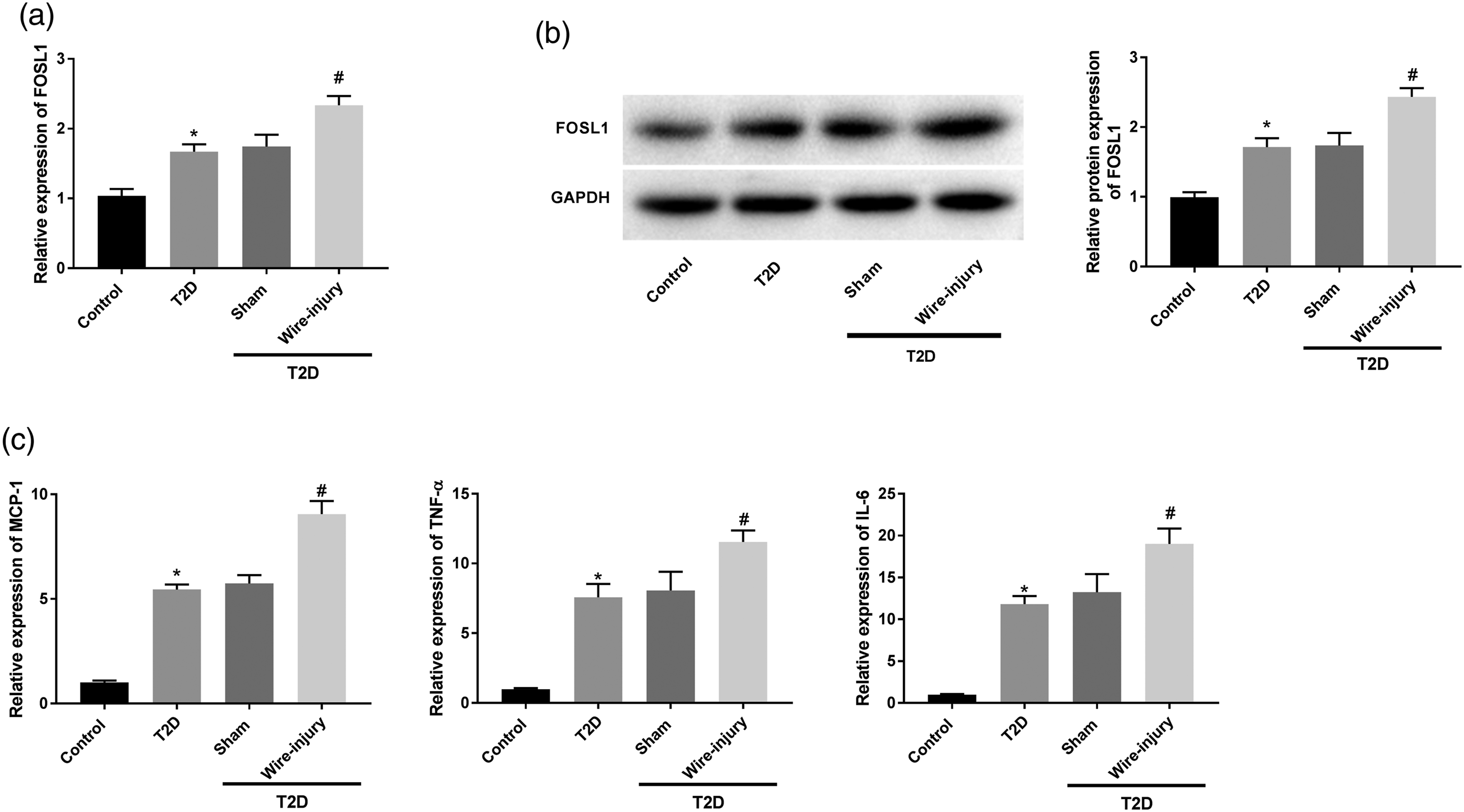
Knockdown of FOSL1 inhibits the viability, migration and inflammation, and promotes the apoptosis of HG-induced VSMCs
We explored the possible role of FOSL1 in HG-induced VSMCs in vitro. As shown in Figure 2(a), the results of qRT-PCR demonstrated that FOSL1 expression was upregulated in HG-treated VSMCs in contrast to that of the NG group (P < 0.05). Thereafter, sh-FOSL1/-NC was transfected into VSMCs to determine the transfection efficiency. Relatively low expression of FOSL1 was detected in VSMCs transfected with sh-FOSL1, suggesting that sh-FOSL1 was transfected into VSMCs successfully (Figure 2(b), P < 0.05). As illustrated in Figures 2(c) to (f), we found that in HG-induced VSMCs, the viability, migration and the levels of MCP-1, TNF-α, and IL-6 were all promoted, but the apoptosis was repressed (P < 0.05). As expected, these were all reversed by FOSL1 silencing (P < 0.05). Knockdown of FOSL1 inhibits the viability, migration and inflammation, and promotes apoptosis in HG-induced VSMCs. (a) The expression of FOSL1 in HG-induced VSMCs was detected by qRT-PCR. *P < 0.05 vs. the NG group. (b) The expression of FOSL1 in VSMCs after transfection with sh-FOSL1/NC was detected by qRT-PCR. *P < 0.05 vs. the sh-NC group. (c) The cell viability of VSMCs after transfection of sh-FOSL1/NC was measured by MTT assay. (d) The migration of VSMCs after transfection of sh-FOSL1/NC was measured by wound healing assay. (e) The apoptosis of VSMCs after transfection of sh-FOSL1/NC was measured by flow cytometric analysis. (f) The mRNA expression levels of MCP-1, TNF-α, and IL-6 in VSMCs after transfection of sh-FOSL1/NC were detected by qRT-PCR. *P < 0.05 vs. the NG group. #P < 0.05 vs. the HG + sh-NC group.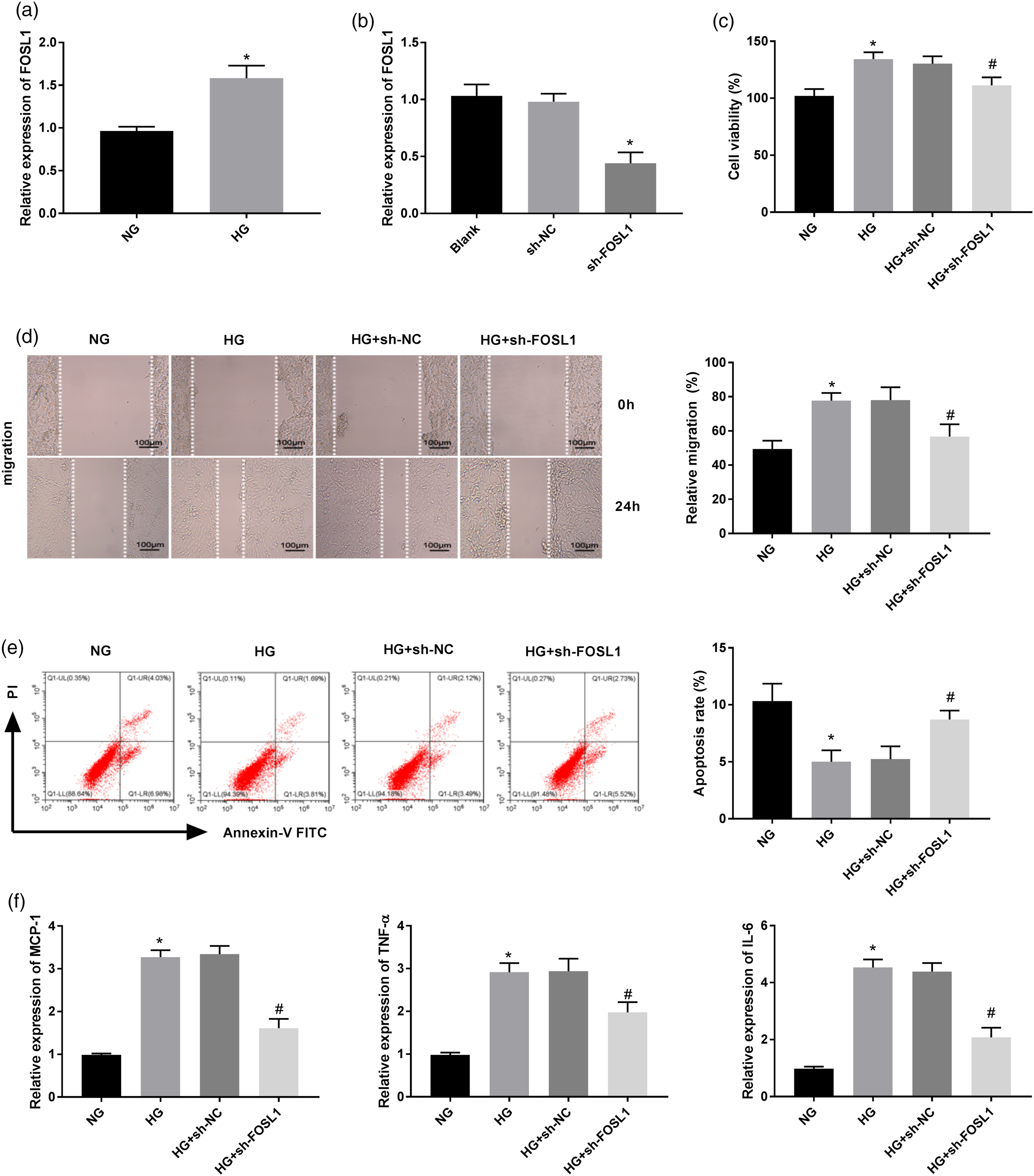
FOSL1 silencing suppresses the ERK/AP-1 pathway in HG-induced VSMCs
It is noticed that ERK/AP-1 pathway is closely associated with cancer proliferation, metastasis, and apoptosis.
13
Therefore, we further explored the interactions between FOSL1 and ERK/AP-1 pathway-related proteins (p-ERK, ERK, c-fos, and c-jun) in HG-induced VSMCs. As presented in Figures 3(a) and (b), HG treatment significantly elevated the protein levels of p-ERK/ERK, c-fos, and c-jun, suggesting that ERK/AP-1 pathway can be activated by HG (P < 0.05). However, these ERK/AP-1 pathway-related protein levels were all repressed by FOSL1 knockdown (P < 0.05). FOSL1 silencing suppresses the ERK/AP-1 pathway in HG-induced VSMCs. (a) The western blot assay images for the levels of p-ERK, ERK, c-fos, and c-jun in VSMCs. (b) The protein levels of p-ERK, ERK, c-fos, and c-jun in VSMCs were measured by western blot assay. *P < 0.05 vs. the NG group. #P < 0.05 vs. the HG + sh-NC group.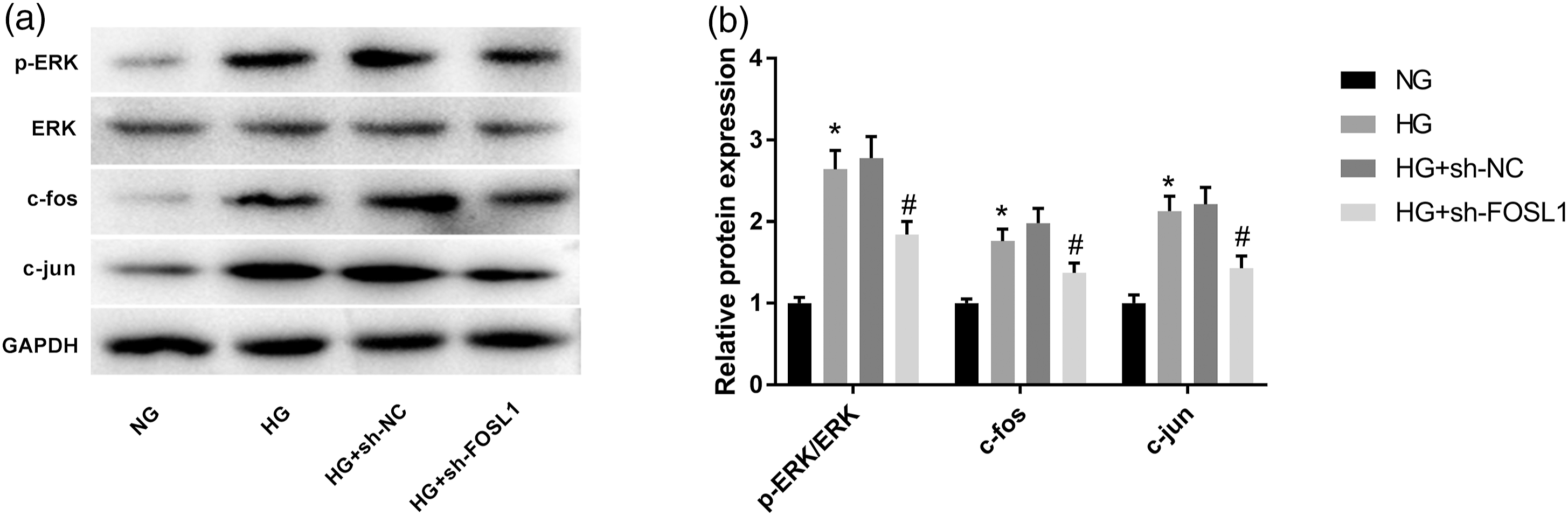
Inhibition of ERK/AP-1 pathway restrains the viability, migration and inflammation, and facilitates apoptosis of VSMCs under HG treatment
U0126 (10 μmol/L), a specific inhibitor of ERK, was added into HG-induced VSMCs to assess the effects of ERK/AP-1 inhibition on cellular processes. In HG-induced VSMCs, addition of U0126 not only decreased cell viability, and migration and inflammatory reactions but also accelerated apoptosis (Figures 4(a) to (d), P < 0.05). Inhibition of ERK/AP-1 pathway restrains the viability, migration and inflammation, and facilitates apoptosis of VSMCs under HG treatment. (a) The cell viability of VSMCs after treatment with U0126 was measured by MTT assay. (b) The migration of VSMCs after treatment with U0126 was measured by wound healing assay. (c) The apoptosis of VSMCs after treatment with U0126 was measured by flow cytometric analysis. (d) The mRNA expression levels of MCP-1, TNF-α, and IL-6 in VSMCs after treatment with U0126 were detected by qRT-PCR. *P < 0.05 vs. the NG group. #P < 0.05 vs. the HG group.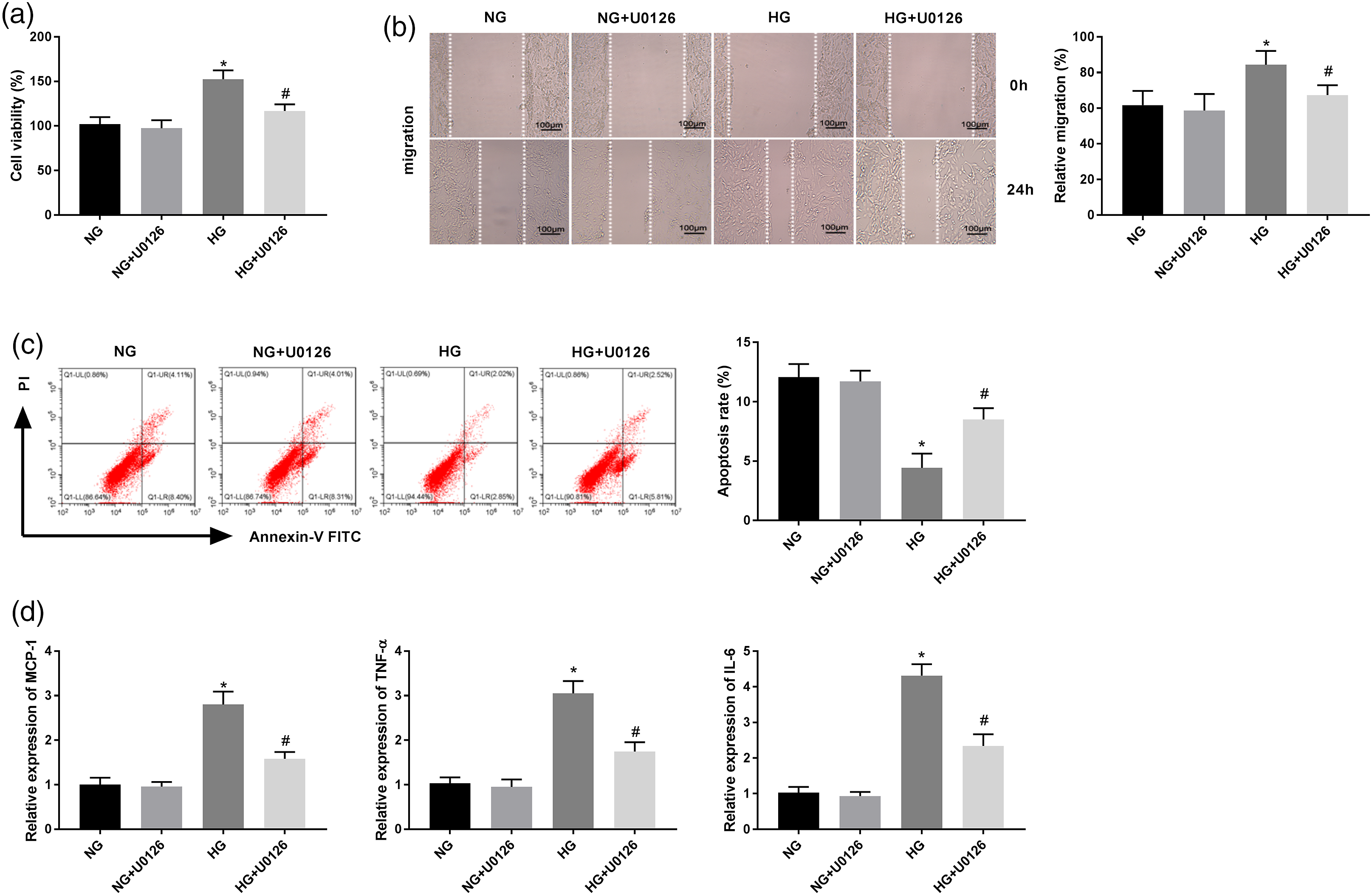
Activation of ERK/AP-1 pathway reverses the effects of FOSL1 knockdown on HG-induced VSMCs
12-O-tetradecanoylphorbol-13-acetate (TPA; 150 nM) was used to activate ERK/AP-1 pathway to perform rescue experiments. As expected, TPA treatment reversed the inhibitory effects of FOSL1 silencing on cell viability, migration and inflammation, and the promoting effect on apoptosis in HG-stimulated VSMCs (Figures 5(a) to (d), P < 0.05). Activation of ERK/AP-1 pathway reverses the effects of FOSL1 knockdown on HG-induced VSMCs. (a) The cell viability of VSMCs after transfection of sh-FOSL1/NC and TPA treatment was measured by MTT assay. (b) The migration of VSMCs after transfection of sh-FOSL1/NC and TPA treatment was measured by wound healing assay. (c) The apoptosis of VSMCs after transfection of sh-FOSL1/NC and TPA treatment was measured by flow cytometric analysis. (d) The mRNA expression levels of MCP-1, TNF-α, and IL-6 in VSMCs after transfection of sh-FOSL1/NC and TPA treatment were detected by qRT-PCR. *P < 0.05 vs. the HG + sh-NC group. #P < 0.05 vs. the HG + sh-FOSL1 group.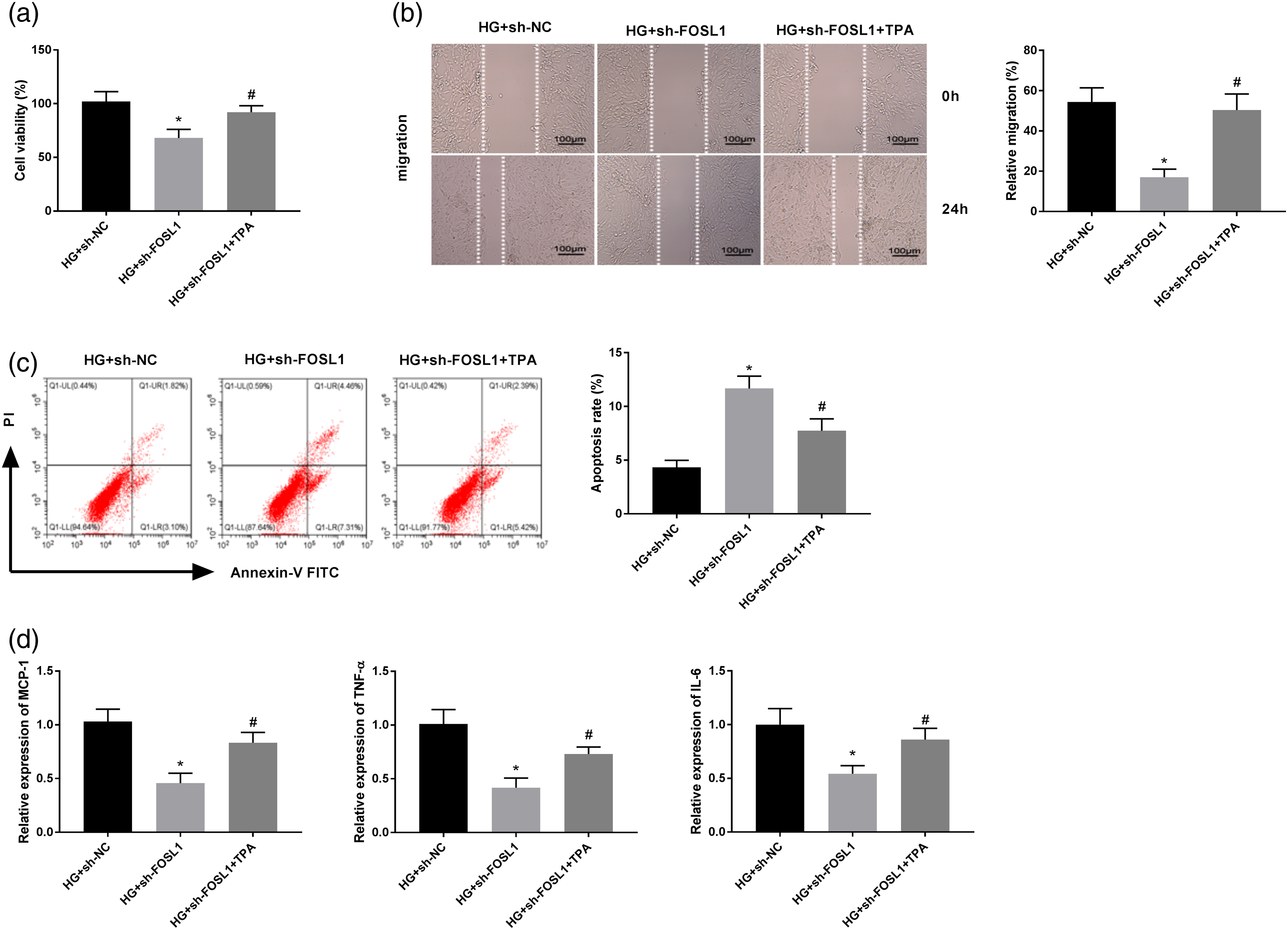
Decreased FOSL1 suppresses neointima formation and inflammation in T2D mice after wire-injury
After injection of Ad-sh-FOSL1, the protein level of FOSL1 in mouse injured common femoral artery was determined by western blotting. We demonstrated that FOSL1 protein level was reduced in the Ad-sh-FOSL1 group compared with the Ad-sh-NC group, which implied that Ad-sh-FOSL1 was injected into wire-injured T2D mice successfully (Figure 6(a), P < 0.05). Afterwards, the histopathological changes of the injured common femoral artery were analyzed using H&E staining. As illustrated in Figure 6(b), the formation of neointima was significantly reduced in the Ad-sh-FOSL1 group compared to that of Ad-sh-NC group. Meanwhile, the areas of neointima and media layers for each section were further calculated. We found an approximately 60% reduction on the area ratio of intima/media caused by FOSL1 knockdown as compared with the corresponding controls (Figure 6(c), P < 0.05). In addition, we also demonstrated that silencing of FOSL1 remarkably repressed the levels of MCP-1, TNF-α, and IL-6 in injured common femoral artery (Figure 6(d), P < 0.05). Decreased FOSL1 suppresses neointima formation and inflammation in T2D mice after wire-injury. (a) The protein level of FOSL1 in T2D mice after wire-injury injected with Ad-sh-FOSL1/NC was measure by western blot assay. (b) The HE staining assay images of T2D mice after wire-injury injected with Ad-sh-FOSL1/NC. (c) The intimal/media ratio of T2D mice after wire-injury injected with Ad-sh-FOSL1/NC was calculated. (d) The mRNA expression levels of MCP-1, TNF-α, and IL-6 in T2D mice after wire-injury injected with Ad-sh-FOSL1/NC were detected by qRT-PCR. *P < 0.05 vs. the Sham group. #P < 0.05 vs. the Ad-sh-NC group.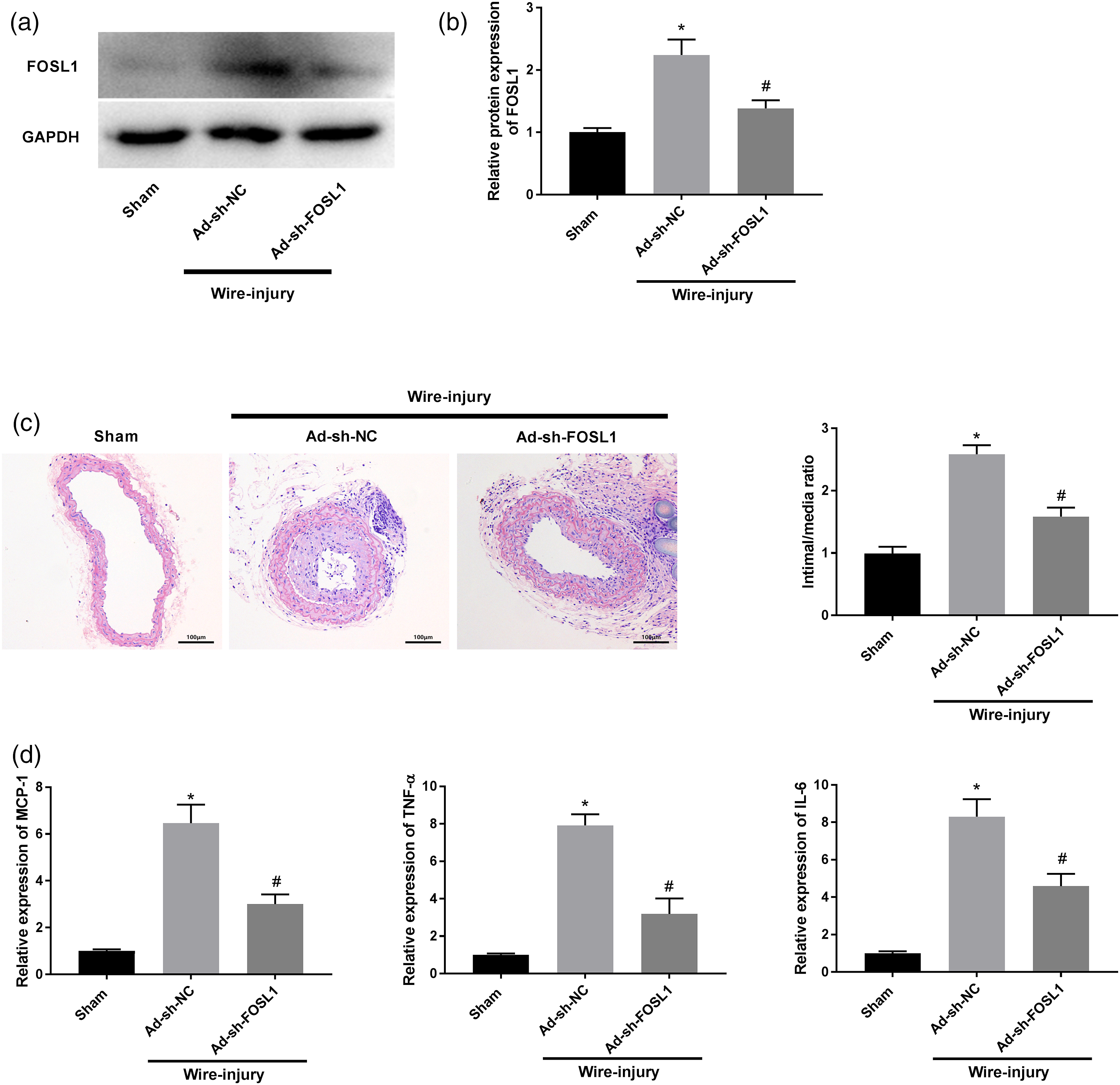
Discussion
Some previous studies have confirmed that hyperproliferative VSMCs and inflammatory reactions are strongly associated with restenosis formation.3,17 Therefore, exploring effective targets to suppress excessive hyperplasia of VSMCs and inflammation are potentially important in the prevention and management of restenosis. In this study, we focused on the detailed effects of FOSL1 on restenosis in vitro and in vivo, and revealed that the ERK/AP-1 pathway may be inactivated by FOSL1 silencing to improve restenosis in T2D mice.
Growing evidence has indicated that the balance of VSMCs viability and apoptosis is important for the occurrence and development of restenosis caused by T2D. 18 Numerous genes or proteins act as important regulators of cellular processes in restenosis.19,20 For instance, Feng et al. 19 observed an increased 14-3-3 gene (YWHAB) in oleic acid–induced VSMCs and believed that the enhanced YWHAB may aggravate restenosis by promoting cell migration. Lv et al. 20 found that Pin1 is upregulated in T2D mice (after wire-injury) tissues and in VSMCs isolated from the above mice, and the further cellular experiments demonstrated that Pin1 downregulation significantly promotes the apoptosis and inhibits metastasis of VSMCs. In this study, relatively high mRNA expression and protein level of FOSL1 was found not only in T2D mice (after wire-injury) tissues but also in HG-treated VSMCs. Besides, silencing of FOSL1 inhibited the cell viability and migration but facilitated the apoptosis of VSMCs treated with HG. In addition, high expression of FOSL1 has been confirmed to be strongly correlated with cell proliferation and metastasis in numerous human cancers.4–6 Based on these results, we speculated FOSL1 may also a pathogenic gene existed in the progression of restenosis. Additionally, inflammation responses are also occurs in the processes of restenosis formation. 21 Therefore, the effects of FOSL1 knockdown on the levels of inflammatory cytokines (MCP-1, TNF-α, and IL-6) were further explored. As expected, we found knockdown of FOSL1 repressed the mRNA expression levels of MCP-1, TNF-α, and IL-6 both in vitro and in vivo, suggesting that FOSL1 silencing plays an important role to suppress the inflammation occurred in restenosis. Vascular neointimal formation is the basis of restenosis and the pathological token of VSMCs hyperplasia. 22 Based on the experimental data of FOSL1 silencing on cell processes of VSMCs we obtained, we speculated that injection of Ad-sh-FOSL1 may reduce the formation of neointima in wire-injury T2D mice. The data of HE staining assay indicated that an approximately 60% reduction was found on the area ratio of intima/media by FOSL1 downregulation as compared with the corresponding controls further validated our assumption. Combined with all the above results, we believed that silencing of FOSL1 can prevent restenosis by inhibition of VSMCs viability and migration.
Recently, increasing attentions have been paid to the promoting effects of ERK/AP-1 activation on the proliferation, migration, and invasion of cancer cells.10–12 We speculated that there may be also some relationships between this pathway and the cellular processes of VSMCs. In this study, we found that U0126 repressed the viability and migration but promoted the apoptosis of HG-induced VSMCs. Similarly, Peng et al. 14 focused on the influence of ERK/AP-1 pathway on VSMCs metastasis in abdominal aortic aneurysm and uncovered that inhibition of ERK/AP-1 pathway remarkably suppressed the motility of VSMCs. Furthermore, reduction of inflammation reactions was also observed in the HG + U0126 group. The results implied that ERK/AP-1 pathway may be also be involved in the progression of restenosis. Given that FOSL1 silencing could inhibit the viability and migration of VSMCs, an assumption that FOSL1 may directly interact with ERK/AP-1 pathway to regulate restenosis was conducted. The results of western blotting indicated that ERK/AP-1 pathway-related proteins (p-ERK/ERK, c-fos, and c-jun) were restrained by FOSL1 knockdown, suggesting that FOSL1 may be positively associated with ERK/AP-1 pathway. Our feedback verification experiments that TPA treatment reversed the inhibiting effects of FOSL1 downregulation on cell viability, migration and inflammatory cytokines, and the promoting effect on apoptosis in HG-induced VSMCs further validated our assumption. Collectively, we indicated that silencing of FOSL1 may repress the viability, migration and inflammation, and accelerate apoptosis of HG-treated VSMCs via the ERK/AP-1 pathway.
Conclusion
In conclusion, this study reveals that FOSL1 knockdown interacts with the ERK/AP-1 pathway to protect against restenosis at a cellular level. However, this study did not confirm the relationship between FOSL1 and ERK/AP-1 pathway in vivo, which may be a limitation. We will consider this in future studies. In summary, we hope these findings provide information on a potential target for the prevention of restenosis in T2D patients underwent PCI.
Footnotes
Author contributions
Conceptualization: Chaoxi Zhou and Haixia Ding.Methodology: Haixia Ding and Hongfang MA.Formal analysis: Fujun Wang and Yaping Du.Investigation: Na Xing and Lin Hou.Writing—original draft preparation: Chaoxi Zhou.Writing—review and editing: All authors.Funding acquisition: Chaoxi Zhou.Resources: Yaping Du.Supervision: Haixia Ding.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.
Research ethics
The procedures of animal experiments in this study were in strict accordance with the Guide for the Care and Use of Laboratory Animals and approved by the ethical committee of The Fourth Hospital of Hebei Medical University.