
Abstract
An emerging body of evidence consistently suggests that compromised blood–brain barrier integrity may be causally associated with cognitive decline induced by type-2 diabetes. Our previous studies demonstrated that selected anti-inflammatory/anti-oxidative agents can preserve the integrity of blood–brain barrier and prevent neuroinflammation in mouse models of dysfunctional blood–brain barrier. Therefore, we have tested whether the previously proven blood–brain barrier protective agent, probucol, can prevent blood–brain barrier breakdown and cognitive decline in a dietary-induced murine model of diabetic insulin resistance. After 6-month chronic ingestion of a diet high in fat and fructose, the mice became insulin resistant. The high-fat and high-fructose-fed mice showed significant cognitive decline assessed by Morris water maze, concomitant with significant elevations in cortical and hippocampal glial acidic fibrillary protein and Fluoro Jade-C staining, indicating heightened neuroinflammation and neurodegeneration, respectively. The integrity of blood–brain barrier in high-fat and high-fructose-fed mice was substantially compromised, and this showed a significant association with heightened neurodegeneration. Co-provision of probucol with high-fat and high-fructose diet completely prevented the cognitive decline and blood–brain barrier dysfunction. Similarly, metformin was able to restore the cognitive function in high-fat and high-fructose-fed mice, while its blood–brain barrier protective effects were modest. These data suggest that probucol may prevent cognitive decline induced by insulin resistance by preserving the integrity of blood–brain barrier, whereas metformin’s neuroprotective effects may be mediated through a separate pathway.
Keywords
Introduction
Type-2 diabetes is reported to significantly accelerate cognitive decline and increase the risk of dementia.1–3 Consistently, animal models of insulin resistance present substantial deterioration in cognitive function.4,5 Although the mechanistic link between diabetes and cognitive decline is not fully understood, evolving evidence suggests that compromised cerebrovascular integrity and function may be centrally involved in insulin resistance–associated cognitive decline.5,6 Min et al. 7 showed that the permeability of blood–brain barrier (BBB) is significantly increased and is associated with cognitive impairment in a mouse model of diabetic insulin resistance. Consistent with this finding, our laboratory recently demonstrated BBB dysfunction preceded neurodegeneration and cognitive decline in a murine model of dietary-induced insulin resistance, suggestive of a causal association. 8 These data collectively suggest that preservation of BBB integrity and function may potentially be an effective therapy to eradicate cognitive deficits in insulin resistance.
Studies using anti-diabetic drugs report inconsistent results on cognitive function in diabetic insulin resistance. Kariharan et al. 9 found that in insulin-resistant db/db mice, the provision of peroxisome proliferator-activated receptor (PPAR)-γ receptor activator, rosiglitazone, improved cognitive performance. Similarly, pioglitazone was demonstrated to reverse memory impairment in a dietary-induced diabetic mouse model. 10 Conversely, however, a population study in ACCORD-MIND (Action to Control Cardiovascular Risk in Diabetes-The Memory in Diabetes) cohort revealed that glitazone treatment exacerbates cognitive decline in some patients with diabetes. 11 In addition, in a study using a dietary-induced insulin resistance mouse model, metformin showed no significant positive effects on cognitive performance, despite the compound improving insulin resistance. 4 Along with the latter findings, a recent systematic review concluded that the insulin-sensitizing treatment of type-2 diabetes does not effectively prevent cognitive impairment and dementia. 12 Importantly, however, none of these studies had a focus on BBB integrity in association with cognitive deficits induced by insulin resistance.
Recent studies demonstrated that nutraceutical and pharmacological agents that present vascular-focussed, anti-inflammatory and anti-oxidative properties can effectively prevent BBB dysfunction.13,14 Previously, our laboratory demonstrated that a lipid-lowering agent with potent anti-oxidative and vascular protective properties, probucol, prevented the increase of BBB permeability and neuroinflammation in high-fat-induced model of BBB dysfunction.15,16 In agreement with our hypothesis, pre-clinical studies reported that probucol mitigates cognitive and hippocampal biochemical changes in mouse models of cognitive impairment.17,18 Therefore, the aim of this study was to investigate whether probucol prevents BBB dysfunction and cognitive decline in an established mouse model of dietary-induced insulin resistance. We also compared the effects of probucol with the most widely used anti-diabetic agent, metformin, because the effects of metformin on BBB in association with cognitive impairment have never been reported. The findings of this study may provide new mechanistic insights into the development and prevention of diabetes-induced cognitive deficits and may offer effective therapeutic opportunities.
Materials and methods
Animals and intervention
Male 6-week-old C57BL/6J mice were purchased from Animal Resources Centre (WA, Australia). A total of 10 mice were randomly allocated to one of the following groups: control, insulin resistant, probucol and metformin. The control group mice received standard maintenance chow containing 4% (w/w) unsaturated fats (AIN93M, Specialty Feeds, WA, Australia). The mice in insulin-resistant group were maintained on a high-fat and high-fructose (HF) diet containing 30% (w/w) lard, 0.5% (w/w) cholesterol and 15% fructose (SF14-088, Specialty Feeds, see Supplementary Tables 1 and 2 for detail). Probucol and metformin groups received the HF diet supplemented with probucol and metformin, respectively. Probucol and metformin were incorporated in diets at 1% (w/w) or 0.2% (w/w), respectively. After 6 months from the commencement of the dietary/pharmacological intervention, all mice were cognitively assessed followed by sacrifice for sample and data collection. Mice were housed in individually ventilated cages under 12-h light/dark cycle in a temperature- and pressure-controlled environment. All mice had ad libitum access to the feed and water. All animal experiments described in this study were approved by Curtin University’s Animal Ethics Committee (approval number AEC_2013_23).
Morris water maze
Short-term spatial learning and memory were tested before sample collection using Morris water maze as detailed previously.7,17 Briefly, the mice first went through 2 days of maze training with visible escape platform (1 cm above water surface), and the mice were trained until they reached the platform within 10 s. The actual maze tests were performed with an invisible platform which was submerged 1 cm below the water surface over 5 consecutive days. Each test day consisted of four trials and maximum time allowed per trial was 90 s. Mice that did not reach the platform within the 90 s were guided to the platform by an investigator. The latency to reach the platform and swim speed was recorded with HVS Image software (HVS Image, UK). The average latency of four trials was calculated and plotted for each day. There was no significant difference in the swim speed between each of the groups (see Supplementary Figure S1).
Brain tissue and plasma sample collection
After the maze test, the mice were anaesthetized with isoflurane gas and blood samples were collected through cardiac puncture into an EDTA-coated tube. Plasma was obtained by centrifugation and stored at −80°C until next use. Brain specimens were removed carefully and washed in phosphate-buffered saline (PBS). The left brain hemisphere was collected into Flow Cytometry (FACs) buffer for flow cytometry analyses (see below for detail), and the right brain hemisphere was collected in 4% paraformaldehyde. The right hemisphere was then cryoprotected in 20% sucrose and frozen in dry ice/isopentane. The tissues were stored at −80°C until next use.
Plasma biochemistry measurements
Blood plasma glucose and HbA1c were measured using AccuCheck Go glucometer (Roche Laboratories, Switzerland) and DCA Vantage Analyzer with DCA HbA1c cartilage (Siemens Healthcare Diagnostics, USA), respectively. Plasma insulin concentration was determined with an enzyme-linked immunosorbent assay (ELISA) kit according to the manufacturer’s instructions (Mercodia, Sweden). Homeostatic model assessment index for insulin resistance (HOMA-IR) was calculated with HOMA Calculator (Diabetes Trials Unit) based on the blood glucose and insulin data.
Plasma concentration of cholesterol was measured using colorimetric kit according to the manufacturer’s instruction (Randox, UK). Briefly, 2 µL of plasma or standard was incubated with 200 µL of reaction solution for 5 min at 37°C. The absorbance was read at 550 nm, and the cholesterol concentrations in samples were extrapolated against a standard curve.
Plasma levels of tumour necrosis factor-alpha (TNF-α), interleukin (IL)-1β, IL-6 and interferon gamma (IFN-γ) were determined using a commercial cytometric bead array method according to the manufacturer’s manual (BD Scientific, Sydney, Australia). Briefly, 50 µL of capture beads mixture for IL-1β, IL-6, TNF-α and IFN-γ; 50 µL of detection reagent; and 50 µL of samples/standards were mixed and incubated at room temperature for 2 h. The beads sandwich was washed with 1 mL wash buffer and resuspended in 300 µL of wash buffer. The data were acquired on BD FACS Canto II and analysed with FCAP Array software.
Assessment of BBB integrity
The integrity of BBB was determined by a semi-quantitative immunomicroscopy measurement of the brain parenchymal extravasation of plasma IgG and flow cytometric measurement of the expression of BBB endothelial tight junction proteins, occludin-1 and ZO-1, as previously established in our laboratory and others.5,19
Briefly, for IgG extravasation measurement, coronal 20 µm cryosections were prepared from the fixed frozen right brain hemisphere. After rehydration and blocking, the sections were incubated with goat anti-mouse IgG conjugated with Alexa488 (1:100, Invitrogen, USA) for 20 h at 4°C. DAPI (4′, 6-diamidino-2-phenylindole) counterstaining was used for the nuclei staining. In order to generate statistically unbiased and sufficiently powered data, between 25 and 40 confocal 3D images were captured within the cortex and hippocampal regions by a blinded experimenter with Ultraview spinning disc confocal microscopy (20× objective, 21 Z-stacks, 1000 × 1000 pixel, 346 × 346 µm) coupled with Volocity software (PerkinElmer, UK). Voxel intensity of the fluorescent dye indicating peri-vascular extravasation of IgG was measured by selecting the region of interest using automatic intensity threshold-based object selection function of Volocity. The accuracy of the automatic selection was manually confirmed by an experienced investigator. Mean voxel intensity was calculated per volume unit per animal and then the group mean was calculated.
Briefly, for flow cytometric analysis of BBB tight junction proteins, the fresh left hemispheres collected in FACs buffer were incubated in a digestion buffer containing 1 mg/mL collagenase IV (Sigma-Aldrich, Missouri, USA), 1 mg/mL dispase (Sigma-Aldrich), 1 mg/mL DNase (Sigma-Aldrich) and 2.5 µg/mL brefeldin for 30 min at 37°C to obtain single floating cells. The samples were subsequently incubated with anti-CD31-BV421 (1:200, Biolegend, CA, USA) and anti-CD45-PerCP-Cy5.5 (1:500, Biolegend). For intracellular marker staining, cells were permeabilized with 0.1% saponin (Sigma-Aldrich) for 15 min and incubated with rabbit anti-mouse occludin-1 (1:50, Abcam, UK) or rabbit anti-mouse ZO-1 (1:100, Abcam) for 30 min at 4°C followed by an incubation with goat anti-rabbit IgG Alexa488 (1:500, LifeTechnologies, Massachusetts, USA) for 30 min at 4 C. Samples were then acquired on FACS Canto II (BD Biosciences, NJ, USA). Cerebrovascular endothelial cells were gated by CD31 positive CD45 negative as reported previously. 19 In order to evaluate the protein expression of interest per endothelial cell, the mean fluorescent intensity of occludin-1 and ZO-1 was analysed with FlowJo V10 software (Treestar, OR, USA) and expressed as per endothelial cell.
Measurement of astrocyte activation and neurodegeneration
The cortical and hippocampal expression of glial fibrillary acidic protein (GFAP) and Fluoro Jade-C staining were measured by semi-quantitative fluorescent microscopy to assess the astrocyte activation and neurodegeneration as described previously. Briefly, 20 µm cryosections of brain right hemisphere were incubated with rabbit anti-mouse GFAP (1:200, Abcam) for 20 h at 4°C. The sections were incubated with goat anti-rabbit IgG conjugated with Alexa488 for 2 h at 20°C. Following the dehydration of sections for 10 min at 60°C, the samples were incubated with sodium hydroxide (Solution A of Fluoro Jade-C kit, Biosensis, SA, Australia) for 5 min. The sections were then incubated with potassium permanganate (solution B) for 10 min, followed by an incubation with Fluoro Jade-C (Solution C) and DAPI (Solution D) for 10 min. Confocal image capture and semi-quantitative analysis of GFAP and Fluoro Jade-C were done as described in the above section for IgG extravasation analysis.
Statistical analyses
Data were calculated and expressed as mean ± SEM. All data in this study were normally distributed, assessed by D’Agostino-Pearson omnibus normality test. Statistical significance was analysed by one-way analysis of variance (ANOVA) followed by Fisher’s least significant difference (LSD) test for multiple comparison. Pearson’s correlation coefficient was used to analyse the associations between the BBB integrity (IgG extravasation) versus neuroinflammation (GFAP expression) and neurodegeneration (Fluoro Jade). The effects of HF diet feeding were considered by analysing the correlation between low-fat (LF) and HF groups, while the effects of probucol or metformin were considered by analysing the correlation between HF and probucol or metformin groups, respectively.
Results
Mice maintained on HF diet for 6 months became insulin resistant, while probucol and metformin improved the insulin sensitivity
The mice fed HF diet for 6 months gained significantly greater weight compared to healthy control mice that received standard LF diet as shown in Figure 1(a). Concomitantly, blood glucose and HbA1c were significantly elevated in HF-fed mice compared to the LF control mice, indicating hyperglycaemia (Figure 1(b) and (c)). Furthermore, the ELISA results revealed that the plasma insulin levels in HF mice were significantly increased compared to LF mice (Figure 1(d)). Based on these findings, HOMA-IR index in mice maintained on HF diet was >threefold higher than the healthy control mice maintained on LF diet (Figure 1(e)), indicating pre-diabetic insulin resistance in HF-fed mice. Plasma concentration of cholesterol was also significantly increased in HF mice compared to LF mice (Figure 1(f)).
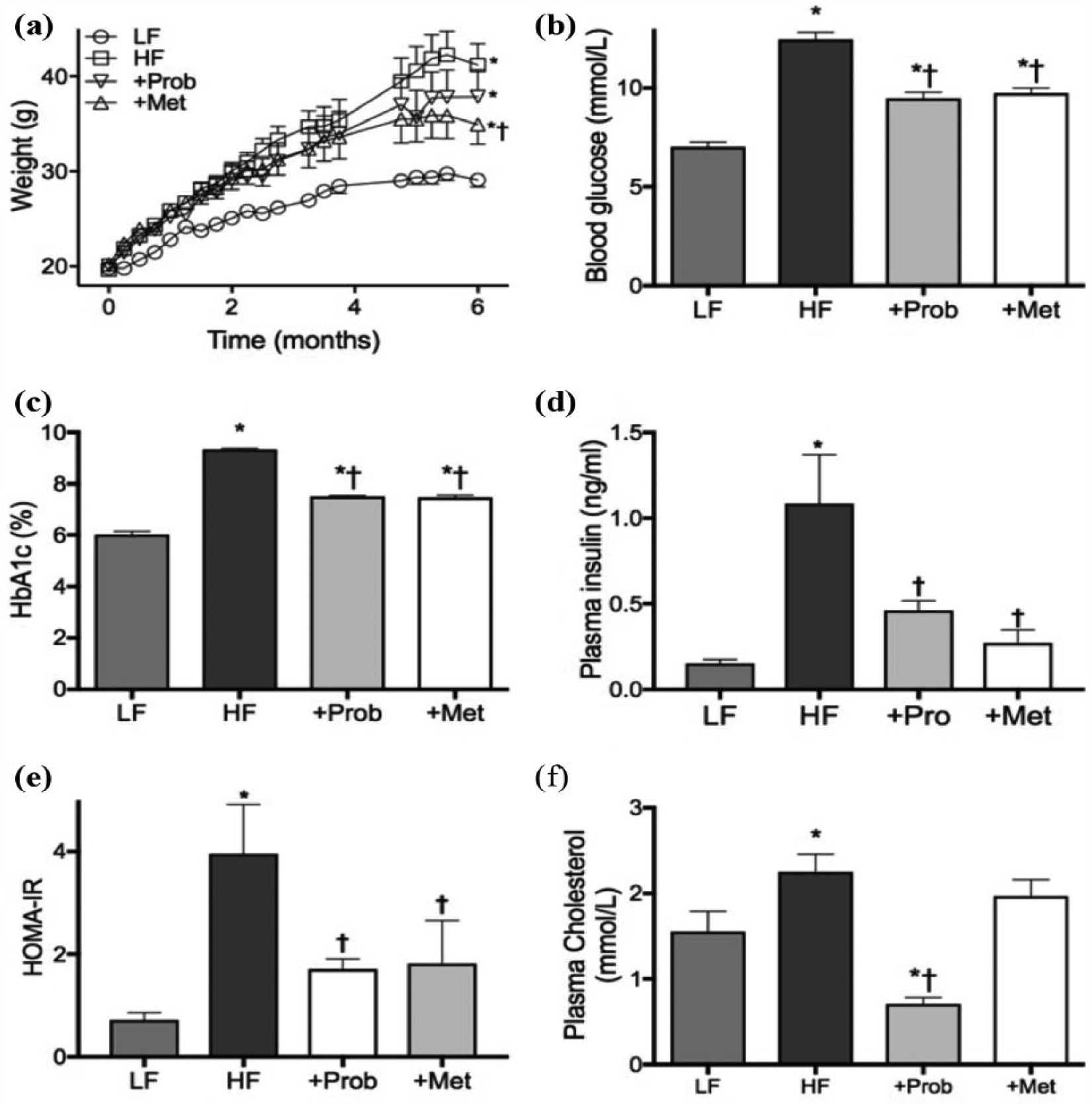
Weights and plasma insulin resistance indicators. (a) The weights of mice that were maintained on low-fat control chow (LF), high-fat and high-fructose diet (HF), HF diet supplemented with probucol (+Prob) or HF diet supplemented with metformin (+Met) for 6 months are presented. (b) Blood glucose levels of LF, HF, +Prob and +Met mice were measured through tail vein before the sample collection. (c) Plasma levels of HbA1c were measured using Siemens DCA Vantage Analyzer. (d) Plasma insulin concentrations were determined using commercial ELISA kit. (e) Based on the glucose, HbA1c and insulin data, homeostatic model assessment index for insulin resistance (HOMA-IR) was estimated using HOMA calculator. (f) Commercial colorimetric assay was used to determine the concentrations of plasma cholesterol. Data are expressed as mean ± SEM. Statistical significance was estimated by one-way ANOVA followed by Fisher’s LSD post hoc test (n = 10, p < 0.05, *significance vs LF; †significance vs HF).
Co-provision of probucol with HF diet had no significant effects on weight gain and the final weight remained significantly higher than the LF control mice (Figure 1(a)). Probucol also showed significant effects in attenuating blood glucose and HbA1c compared to the HF-fed mice (Figure 1(b) and (c)); however, both markers remained significantly higher than the healthy control mice but within the range of normoglycaemia. Nonetheless, probucol completely normalized plasma insulin levels, and thus, the HOMA-IR index of mice co-supplemented with probucol was comparable to healthy, non-insulin-resistant LF mice (Figure 1(e)). Probucol also showed remarkable effects in reducing plasma cholesterol, significantly lower than the LF mice (Figure 1(f)).
Co-provision of metformin with HF diet significantly attenuated weight gain compared to the mice that were fed only HF diet; however, the final weight still remained significantly greater than the healthy control mice maintained on LF diet (Figure 1(a)). Similar to probucol, metformin supplementation significantly reduced the blood glucose and HbA1c levels compared to the HF mice, but they both remained significantly greater than the LF control mice (Figure 1(b) and (c)). Nevertheless, plasma insulin levels were significantly attenuated by metformin and the HOMA-IR index was comparable to health LF mice (Figure 1(d) and (e)). Metformin supplementation did not show any significant effects on plasma cholesterol (Figure 1(f)).
Probucol and metformin prevented cognitive deficits, astroglial activation and neurodegeneration in HF-fed insulin-resistant mice
After the 6-month dietary intervention with a pro-diabetic diet enriched in fat and fructose (HF), the insulin-resistant mice showed significantly impaired spatial learning and memory indicated by the latency of Morris water maze, while the healthy control mice maintained on LF diet showed no sign of cognitive decline (Figure 2(a) and (b)). Concomitantly, semi-quantitative immunomicroscopy analysis showed significantly elevated astroglial activation (indicated by GFAP) and neurodegeneration (indicated by Fluoro Jade-C) in the cortex and hippocampal formation of HF-fed mice compared to the LF control mice (Figure 2(c) to (e)).
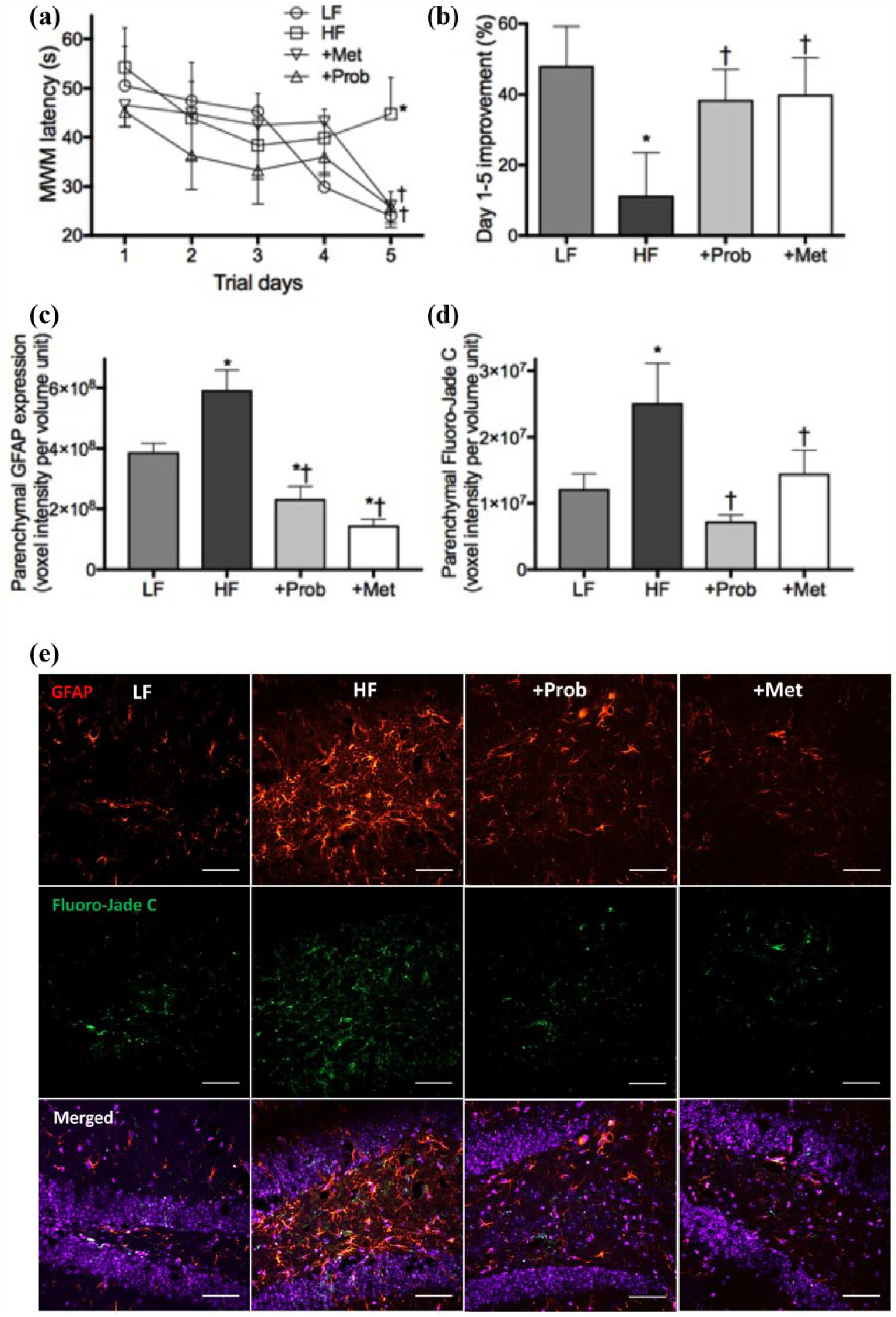
Cognitive function and neurodegeneration. (a) Hippocampal-dependent spatial learning and memory were assessed using Morris water maze, and the mean latency to reach escape platform of mice maintained on low-fat control chow (LF), high-fat and high-fructose diet (HF), HF diet supplemented with probucol (+Prob) or HF diet supplemented with metformin (+Met) for 6 months are presented. (b) Improvement of the escape latency from Day 1 to Day 5 of each group is expressed as per cent. (c and d) Semi-quantitative confocal immunomicroscopy was used to determine the astrocyte activation indicated by glial acidic fibrillary protein (GFAP) and the neurodegeneration indicated by Fluoro Jade-C staining. (e) Representative immunomicrographs of GFAP (red) and Fluoro Jade-C (green) in hippocampal dentate gyrus are shown with DAPI nuclei counterstaining (blue). The scale bar indicates 50 µm. Data are expressed as mean ± SEM. Statistical significance was estimated by one-way ANOVA followed by Fisher’s LSD post hoc test (n = 10, p < 0.05, *significance vs LF; †significance vs HF).
The co-provision of probucol with HF diet completely prevented cognitive dysfunction, showing the comparable rate of latency improvement from Day 1 to Day 5 to LF-fed healthy control mice (Figure 2(a) and (b)). The increase of GFAP expression and Fluoro Jade-C staining in HF-fed mice were also significantly attenuated by probucol (Figure 2(c) to (e)). Similarly, the spatial learning and memory function of mice supplemented with metformin was comparable to the healthy LF-fed mice (Figure 2(a) and (b)). Consistent with these findings, the expression of GFAP and Fluoro Jade-C staining was completely normalized by metformin (Figure 2(c) to (e)).
HF-induced BBB dysfunction was prevented by probucol but not metformin
The mice maintained on HF diet for 6 months showed significantly elevated parenchymal extravasation of IgG in the cortex and hippocampal formation, while LF-fed healthy control mice exhibited no sign of substantial IgG extravasation (Figure 3(a) and (b)). Consistent with these findings, flow cytometric analysis showed significantly lowered expression of occludin-1 and ZO-1 tight junction protein in the cerebrovascular endothelial cells of HF group mice compared to the healthy control LF mice (Figure 3(c) and (d)). Co-provision of probucol with the HF diet completely prevented parenchymal IgG extravasation, coinciding with restoration of the expression of occludin-1 and ZO-1. In contrast, the BBB protective effects of metformin were not as significant, where parenchymal IgG extravasation remained significantly greater than the LF controls (Figure 3(a)). Indeed, substantial parenchymal IgG extravasation was observed in the cortex of metformin-treated mice (Figure 3(b)). Concomitantly, the expression of BBB tight junction proteins, occludin-1 and ZO-1, in mice co-supplemented with metformin showed no significant difference from the HF-fed mice (Figure 3(c) and (d)).
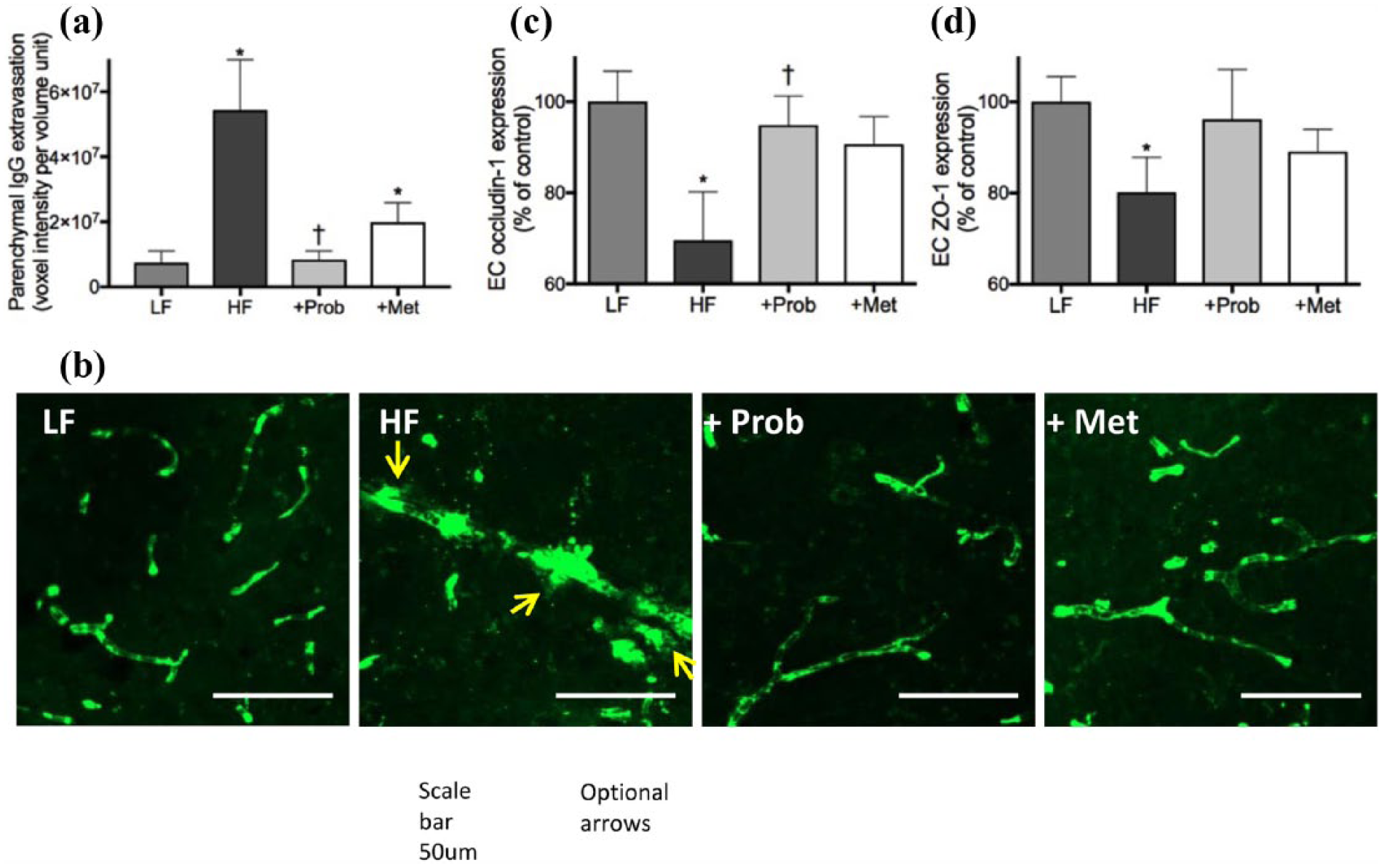
Assessment of blood–brain barrier integrity. The integrity of blood–brain barrier (BBB) was assessed by a semi-quantitative measurement of parenchymal extravasation of plasma IgG, as well as by a flow cytometric analyses of cerebrovascular endothelial expression of BBB tight junction proteins, occludin-1 and ZO-1, in mice maintained on low-fat control chow (LF), high-fat and high-fructose diet (HF), HF diet supplemented with probucol (+Prob) or HF diet supplemented with metformin (+Met) for 6 months. (a) The semi-quantitative data of IgG extravasation in the cortex and hippocampal formation are shown. (b) Representative immunomicrographs of cortical IgG extravasation are presented. IgG is indicated in green and the site of IgG extravasation is indicated with arrows. Scale bar indicates 50 µm. The graphs show semi-quantitative data of endothelial cell expression of occludin-1 (c) and ZO-1 (d) expressed as per cent of control LF mice. Data are expressed as mean ± SEM. Statistical significance was estimated by one-way ANOVA followed by Fisher’s LSD post hoc test (n = 10, p < 0.05, *significance vs LF; †significance vs HF).
HF diet-induced elevation of plasma inflammatory cytokines was significantly suppressed by probucol
Plasma concentration of IL-1β was significantly increased by the ingestion of HF diet compared to the healthy control mice feeding (Figure 4(a)), while plasma IL-6 remained unaffected by the HF feeding (Figure 4(b)). The co-provision of probucol with HF diet significantly inhibited the plasma IL-1β, while no significant effects were observed with IL-6. Similarly, the co-provision of metformin significantly reduced plasma IL-1β, but no effects were seen with IL-6.
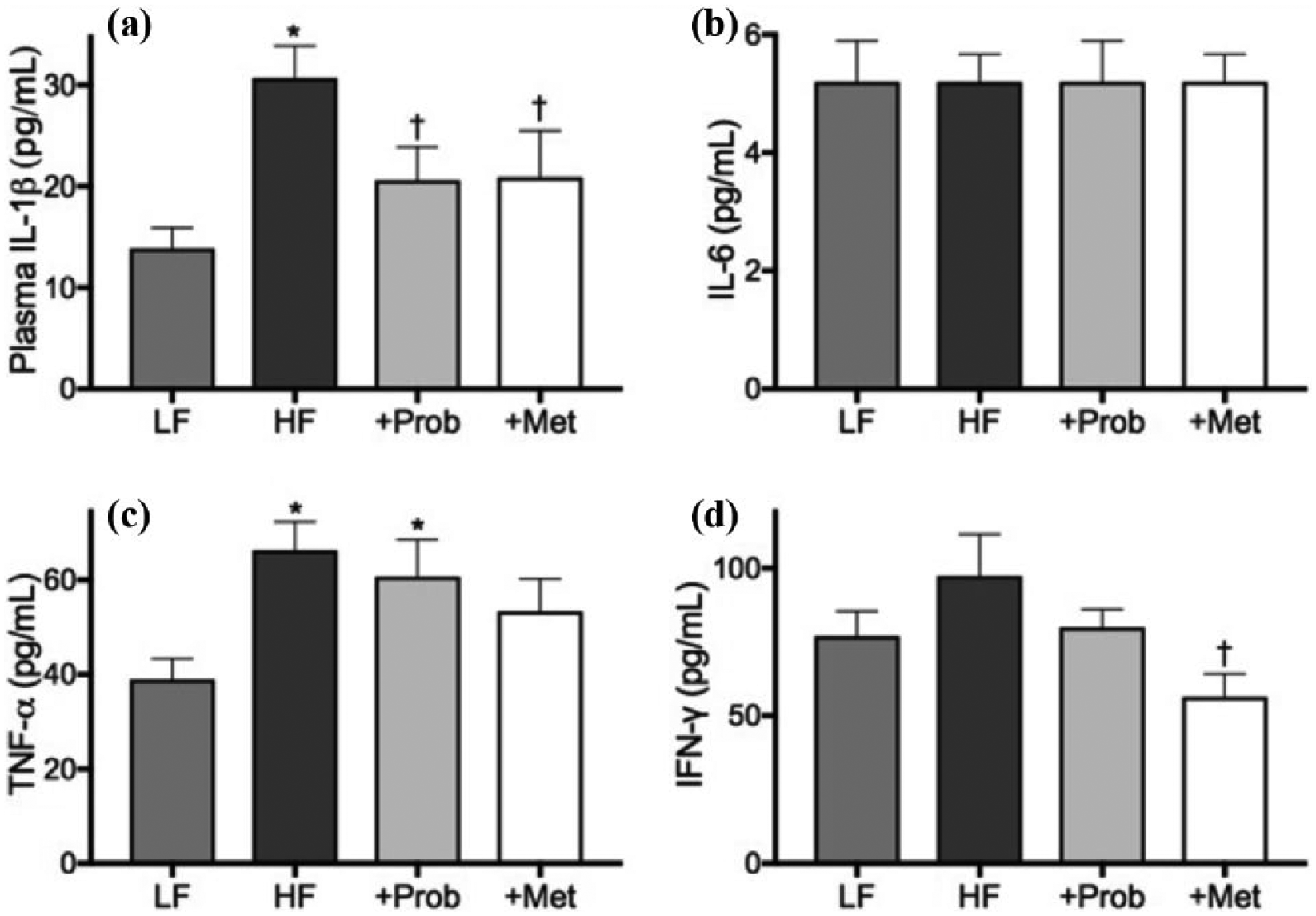
Measurement of plasma pro-inflammatory cytokines. Plasma concentrations of pro-inflammatory cytokines, (a) IL-1β, (b) IL-6, (c) TNF-α and (d) IFN-γ, were measured by commercial cytometric beads array kit in mice maintained on low-fat control chow (LF), high-fat and high-fructose diet (HF), HF diet supplemented with probucol (+Prob) or HF diet supplemented with metformin (+Met) for 6 months. Data are expressed as mean ± SEM. Statistical significance was estimated by one-way ANOVA followed by Fisher’s LSD post hoc test (n = 10, p < 0.05, *significance vs LF; †significance vs HF).
The HF diet also increased the concentration of plasma TNF-α significantly compared to the LF control mice (Figure 4(c)). Probucol and metformin exerted no significant effects on this. The plasma concentration of IFN-γ showed increasing trend in HF-fed mice compared to LF-fed control mice, while probucol and metformin appeared to suppress plasma concentration (Figure 4(d)).
BBB permeability showed significant associations with neuroinflammation and neurodegeneration in probucol-treated mice
Pearson’s correlation coefficient analysis shows significant correlation between the IgG extravasation versus GFAP expression and Fluoro Jade in HF-fed insulin-resistant mice (Figure 5(a) and (d)), indicating the neuroinflammation and neurodegeneration is associated with HF-induced BBB dysfunction. A significant correlation was also indicated between IgG extravasation versus GFAP expression and Fluoro Jade in probucol supplemented mice, suggesting that probucol may attenuate neuroinflammation and neurodegeneration by improving BBB integrity (Figure 5(b) and (d)). In contrast, no significant correlation was indicated in IgG extravasation versus Fluoro Jade in metformin co-supplemented mice (Figure 5(c) and (d)), suggesting that metformin’s neuroprotective effects may not be strongly associated with BBB integrity.
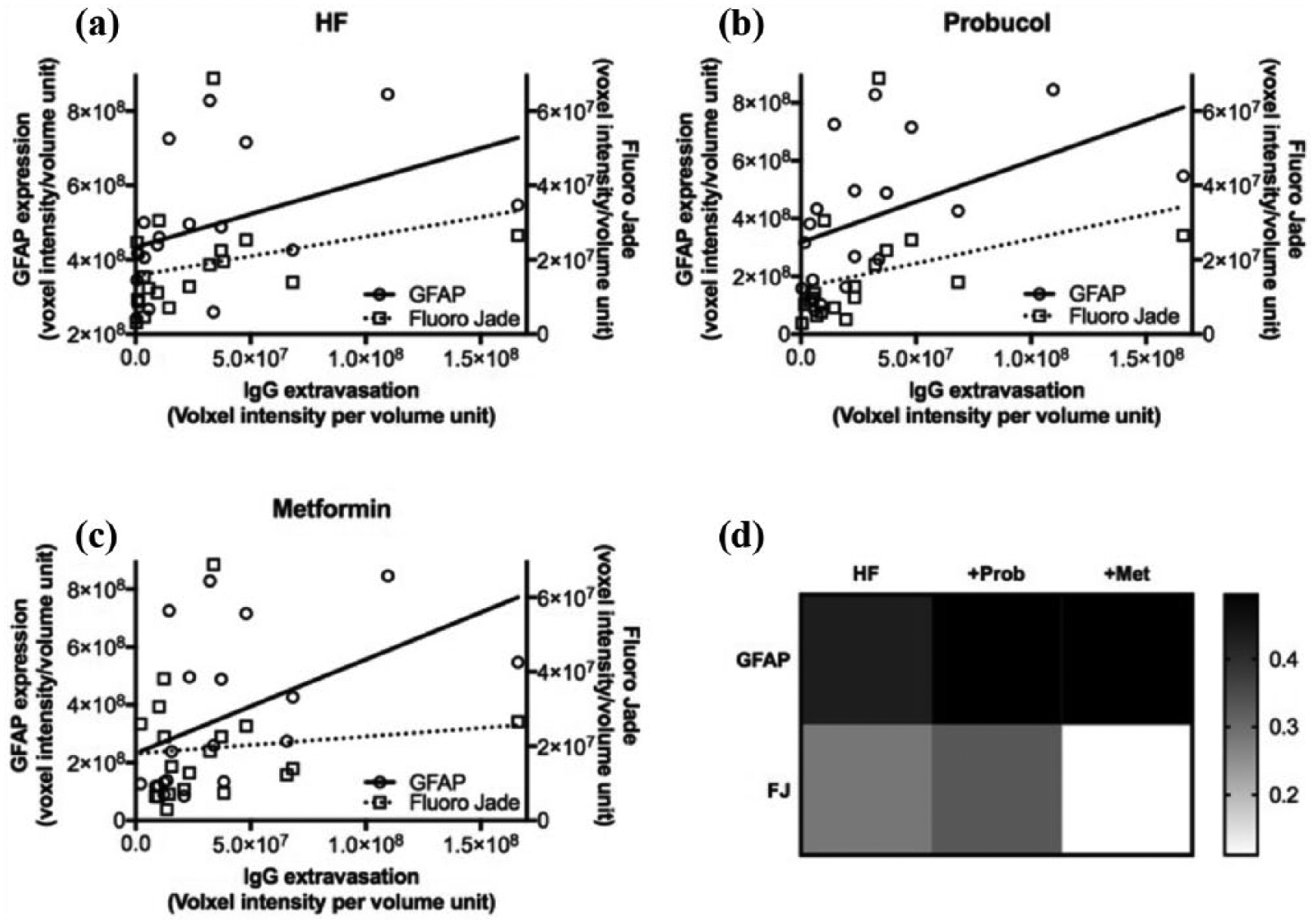
Correlation coefficient analyses of BBB dysfunction with neuroinflammation and neurodegeneration. The associations between BBB dysfunction with neuroinflammation and neurodegeneration were considered with Pearson’s correlation coefficient analysis. (a) The effect of HF dietary intervention was assessed using the data sets of LF and HF mice. (b) The effect of probucol supplementation was assessed using the data sets of HF and probucol group. (c) The effect of metformin was assessed using the data sets of HF and metformin group. (d) The r values of correlation coefficient analyses are summarized and presented in a heatmap.
Discussion
An accumulating body of evidence is consistent with the notion that compromised BBB integrity may be mechanistically involved in the development of cognitive decline in diabetic insulin resistance. This study used a previously established BBB protective drug, probucol, to explore its BBB-modulating properties and cognitive decline in an established dietary-induced insulin-resistant mouse model and investigated whether the protection of BBB can play a role in the prevention of diabetes-induced cognitive decline. In addition, we compared the effects of probucol with anti-diabetic metformin.
Indicated by significant increases in plasma glucose, HbA1c, insulin and HOMA-IR, the mice maintained on HF diet for 6 months were insulin resistant. According to our recently published study, the mice would have been mildly insulin resistant after 4 weeks from the commencement of HF dietary intervention. 8 The quantitative immunofluorescent confocal microscopy analysis indicated substantial cerebral parenchymal extravasation of plasma-derived IgG in the HF-fed insulin-resistant mice, indicating significantly exaggerated BBB permeability compared to the LF control mice. Furthermore, flow cytometric analyses of cerebrovascular endothelial cells confirmed that the insulin resistance–induced BBB disruption was mediated by the decreased expression of endothelial tight junction proteins, occludin-1 and ZO-1. Studies report that the expression of BBB tight junction proteins is highly influenced by inflammatory pathways. IL-1β and TNF-α are reported to degrade the tight junction proteins and dysregulate its cellular translocation, while the inhibition of these cytokines preserves the tight junctions in cerebrovascular endothelial cells.20–23 Consistently, in our study, the plasma concentrations of pro-inflammatory cytokines, IL-1β and TNF-α, were significantly elevated in HF-fed mice compared to the healthy control mice fed with LF diet, while no changes were observed for plasma IL-6 and IFN-γ. These data collectively suggest that in diabetic insulin resistance, elevated TNF-α and IL-1β may be responsible for the dysregulation of BBB endothelial tight junction complex assembly, which in turn leads to the dysfunction of BBB and blood-to-brain unspecific efflux of plasma-borne molecules such as IgG.
In addition to the circulating inflammatory cytokines, other factors may also increase cerebrovascular endothelial inflammation, which may also modulate BBB tight junction protein expression. Excessive cholesterol supply to the endothelial cells may be one of those factors. Cellular cholesterol overload is reported to induce mitochondrial and endoplasmic reticulum stress in endothelial cells. 24 Such mitochondrial activity and lysosomal processing are known to result in increased oxidative stress and inflammation. 25 In concert with this, our findings show that HF mice had significantly elevated plasma cholesterol compared to the LF mice. Studies also suggest that the increased blood glucose may influence the permeability of BBB.26,27 Chronic hyperglycaemia is reported to exaggerate oxidative stress and inflammation through activated receptor for advanced glycation end products (RAGE), which consequentially disrupt BBB. 28 These data collectively suggest that in dietary-induced insulin-resistant mice, the dysfunction of BBB may be mediated by several factors including the heightened circulating pro-inflammatory cytokines, elevated plasma cholesterol and hyperglycaemia.
Consistent with previous studies by others and our laboratory,4,5 the HF-induced insulin-resistant mice exhibited significant decline in hippocampal-dependent spatial learning and memory tested by Morris Water Maze, while the control LF mice showed healthy cognitive performance over 6 months of intervention. Such findings of cognitive deficits in HF-fed mice were concomitant with substantive indications of neuroinflammation and neurodegeneration. Moreover, the significant association observed between neurodegeneration/neuroinflammation and BBB permeability complements the findings from our recent study, which reported on the potential causal link between BBB dysfunction and cognitive decline in dietary-induced insulin-resistant mice. 8
This study demonstrated probucol’s remarkable BBB protective effects preventing the blood-to-brain extravasation of plasma-derived IgG in HF-fed insulin-resistant mice. The flow cytometric analysis of cerebrovascular endothelial cells revealed that the BBB protective effects of probucol were mediated by the restoration of occludin-1 and ZO-1 expression. In addition, probucol significantly suppressed the plasma concentration of IL-1β and cholesterol. Co-supplementation of probucol with HF diet also reduced plasma glucose and HbA1c levels, although these remained slightly higher than the normoglycaemia. These data suggest that probucol may protect BBB integrity in insulin-resistant mice through the suppression of peripheral inflammation and cholesterol oversupply to the cerebrovascular endothelial cells, and to lesser extent, through improving hyperglycaemia.
Studies report that probucol attenuates hippocampal neuroinflammation and oxidative stress, and by extension, prevents deterioration of cognition in animal models of cognitive impairment.17,18 These models included streptozotocin-induced type-1 diabetes, as well as β-amyloid-induced neurodegeneration. However, no studies to date have evaluated the neuroprotective effects of probucol relative to the integrity of BBB. In this study, the escape latency of Morris Water Maze indicated that co-supplementation of probucol with HF diet completely normalized spatial learning and memory. In agreement with this finding, the cerebral cortex and hippocampal neuroinflammation and neurodegeneration were significantly attenuated by probucol compared to the HF-fed insulin-resistant mice. The correlation coefficient analysis between the HF-fed mice and HF + probucol mice revealed that the permeability of BBB significantly correlates with neuroinflammation and neurodegeneration. These data collectively suggest that the prevention of cognitive impairment in HF-induced insulin resistance by probucol may be mediated through the attenuation of BBB disruption.
Metformin is one of the most widely prescribed insulin-sensitizing, diabetes medications. The previous reports on metformin’s neuro- and cognition-protective effects are inconsistent. Furthermore, no previous studies considered the effects of metformin on BBB integrity in association with cognitive decline in diabetes. While our data indicated some trends in preserving BBB structure and function by metformin in HF-induced insulin-resistant mice, the BBB protective effects of metformin were modest compared to probucol. Nonetheless, metformin appeared to suppress inflammation by significantly downregulating peripheral IL-1β and IFN-γ. As expected and similar to the effects of probucol, the co-supplementation of metformin with HF significantly attenuated blood glucose and HbA1c. However, unlike probucol, plasma cholesterol remained unaffected by metformin. These data collectively suggest that the normalization of all three factors – (1) plasma inflammatory cytokines, (2) hyperglycaemia and (3) hypercholesterolaemia – may be essential to completely prevent the BBB dysfunction in diabetic insulin resistance.
Despite its modest BBB protective effects, metformin appeared to significantly suppress neuroinflammation and neurodegeneration and prevented cognitive impairment in HF-induced insulin-resistant mice. This suggests that metformin may ameliorate cognitive decline in diabetic insulin resistance via mechanisms not directly through the BBB axis. In rodent models of neurological dysfunction, studies showed that metformin can cross the BBB and activate adenosine monophosphate–activated protein kinase (AMPK),29,30 indicating that metformin may directly influence central nervous system (CNS) neurons to protect their integrity. Indeed, this is further supported by in vitro studies reporting on the substantial neuroprotective effects of metformin in preventing apoptosis and increasing viability of CNS neuronal cells.31,32 In diabetic insulin–resistant models, metformin is demonstrated to improve neuronal redox imbalance and neuroblast proliferation and differentiation in hippocampal dentate gyrus.33,34 Gupta et al. 35 also reported that metformin improves insulin sensitivity of neurons by promoting the activation of AMPK, PI2 kinase and Akt in insulin-resistant mouse neuroblastoma cell line induced by chronic hyperinsulinaemic condition. However, in this study, such metformin’s direct actions on CNS were not investigated.
This study reports for the first time that probucol and metformin completely prevent cognitive deficits by attenuating the neuroinflammation and neurodegeneration in dietary-induced pre-diabetic insulin-resistant mouse model. Our data also suggest that such neuroprotective effects of probucol and metformin may be at least partially, if not entirely, mediated through their BBB protective properties. While further studies are needed to identify the exact mechanisms of neuroprotective action of each agent, the outcomes may offer important therapeutic opportunities for subjects with diabetic insulin resistance who are at significantly greater risk in developing cognitive impairment and dementia. Future studies may also consider the combination therapies of probucol and metformin or other anti-diabetic drugs to further increase the efficacy of these drugs on BBB and cognitive function. Studies using other models of type-2 diabetes such as db/db mice may also provide useful information.
Footnotes
Acknowledgements
The authors would like to acknowledge Jeanne Le Masurier at Curtin Health Innovation Research Institute and Dr Mina Elahy at University of New South Wales for their technical assistance in collecting the flow cytometric data.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: The study was financially supported by National Health and Medical Research Council of Australia, Alzheimer’s Australia Dementia Research Foundation, Western Australian Department of Health and Curtin University.