
Abstract
Introduction:
Diabetes mellitus appears to be negatively associated with abdominal aortic aneurysm; however, the mechanisms underlying this relationship remain poorly understood. The aim of this article is to provide a comprehensive review of the currently understood biological pathways underlying this relationship.
Methods:
A review of the literature (‘diabetes’ OR ‘hyperglycaemia’ AND ‘aneurysm’) was performed and relevant studies grouped into biological pathways.
Results:
This review identified a number of biological pathways through which diabetes mellitus may limit the presence, growth and rupture of abdominal aortic aneurysms. These include those influencing extracellular matrix volume, extracellular matrix glycation, the formation of advanced glycation end-products, inflammation, oxidative stress and intraluminal thrombus biology. In addition, there is an increasing evidence to suggest that the medications used to treat diabetes can also limit the development and progression of abdominal aortic aneurysms.
Conclusion:
The negative association between diabetes and abdominal aortic aneurysm is robust. Future studies should attempt to target the pathways identified in this review to develop novel therapeutic agents aimed at slowing or even halting aneurysm progression.
Introduction
Diabetes mellitus (DM) is a strong cardiovascular risk factor; however, multiple epidemiological studies have confirmed that a negative relationship exists between DM and abdominal aortic aneurysm (AAA) presence, growth and rupture.1–5 Arteries from patients with DM are often harder and more calcified than those from patients without DM; however, increased vessel wall calcification alone does not appear to explain the reduced rate of aortic expansion seen in diabetic patients. 6
Understanding the biological basis for negative associations between DM and AAA is important since there are currently no medical therapies that can be used to slow or halt the progression of AAA. Two earlier papers7,8 have provided some insight of the mechanisms involved; however, since these were published, there have been numerous other studies which are included in this contemporary review of the literature.
Methods
A MEDLINE search was performed using the terms ‘diabetes’ OR ‘hyperglycaemia’ AND ‘aneurysm’ in December 2017 which identified >1900 papers for review. Title and abstract review narrowed the list of relevant papers down to 31 which then all underwent full-text review. All papers were in the English language. The reference lists of these papers were also searched to identify additional papers of relevance such as those relating to diabetic nephropathy or retinopathy where shared pathogenetic mechanisms might be important. The review detailed below represents the summary of this literature search as well as the authors’ personal knowledge of the subject area.
Results
Although various studies have been published in this area, the results can be broadly grouped into four main biological pathways affecting: (1) extracellular matrix (ECM) volume, (2) ECM glycation and advanced glycation end-product (AGE) formation, (3) inflammation and oxidative stress and (4) intraluminal thrombus (ILT) biology. Significant research also suggests that the medications used to treat DM may play more of a role than the disease itself in explaining this relationship; therefore, the literature in this area has also been reviewed.
ECM volume
Many of the studies assessing the impact of hyperglycaemia on ECM volume have focused on patients with diabetic kidney disease. These studies show that chronic hyperglycaemia is associated with a progressive increase in ECM volume due to a decrease in ECM degradation and an increase in ECM synthesis. 9 In the kidney, this results in the development of diabetic nephropathy; however, in the aorta, this probably results in arterial wall stiffening. 8 Astrand et al. 10 have shown that diabetic patients undergoing ultrasound of their aorta have a significantly thicker intima-media layer when compared to non-diabetic age- and sex-matched controls (0.89 mm vs 0.73 mm; p < 0.001). The same group found that this results in diabetic aortas being subjected to 20% less aortic wall stress (7.8 × 105 dynes/cm2 vs 9.7 × 105 dynes/cm2; p < 0.001) which may be one explanation for the reduced rate of AAA growth among diabetic patients. 3
The metabolic milieu of DM can exert direct effects on the production of ECM proteins. Jones et al. 11 found that 25 mmol/L of intermittent hyperglycaemia was capable of increasing collagen synthesis by 29% in cultured human tubulointerstitial cells. In contrast, Shi et al. 12 found that hyperinsulinaemia, which is often present with hyperglycaemia in patients with insulin resistance, was associated with increased elastin gene expression via a PI3K-dependent pathway in cultured human aortic smooth muscle cells (AoSMCs). This is important since disruption of elastin fibres is one of the key histological hallmarks of AAA. 13
Matrix metalloproteinases (MMPs) are important in the pathogenesis of AAA since they are key regulators of ECM volume and structure. Although results are not uniform in this area, numerous studies have found hyperglycaemia to be associated with a reduced level of MMP expression. Death et al. 14 found that 25 mmol/L of hyperglycaemia was able to down-regulate expression of MMP-3 in endothelial cells; however, expression of MMP-2, one of the key MMPs in pathogenesis of AAA, was up-regulated by hyperglycaemia in the same study. Portik-Dobos et al. 15 used internal mammary artery specimens from patients with and without DM and found significantly decreased levels of pro-MMP-1, pro-MMP-2 and total MMP activity (p < 0.05) in specimens from diabetic patients. In a different study, the same group reported decreased levels of MMP-1 and MMP-9 in internal mammary and crural vessel specimens from diabetic patients undergoing lower limb amputation. 16 In the kidney, DM is associated with reduced MMP-2 expression, 17 mediated through a pathway involving hyperglycaemia-induced angiotensin II production. 18 Angiotensin II in turn stimulates the expression of transforming growth factor (TGF)-β1 which has been shown to down-regulate MMP-2 expression. 19 Plasmin is another regulator of MMP expression since plasmin catalyses the conversion of pro-MMP-2 and pro-MMP-9 to their active forms MMP-2 and MMP-9. Dua et al. 20 have shown using the elastase infusion mouse model of AAA that hyperglycaemia can increase levels of plasminogen activator inhibitor-1 (PAI-1) which ultimately decreases plasmin expression and hence MMP levels.
ECM glycation and AGE formation
Chronic hyperglycaemia is associated with non-enzymatic cross-linking of ECM proteins which results in the formation of so-called AGEs which in vascular tissues can increase vessel stiffness. 21 Chirinos et al. 22 performed pulse wave velocity measurements in patients with and without T2DM and found increased large artery stiffness, decreased reflection magnitude and increased pulsatile energy transmission to distal arterial beds among those with T2DM.
The importance of AGEs in protecting against AAA is complex. Norman et al. reported lower circulating levels of carboxymethyllysine (CML) among diabetic males with AAA compared to those without AAA and hypothesised that increased AGE formation among diabetic patients may explain the protective effect of DM on AAA; however, the study was criticised since CML does not induce cross-linking.23,24 In addition, Zhang et al. 25 found higher tissue concentrations of AGEs in aortic specimens from patients with AAA and suggested that AGEs together with their receptor (RAGE) are important for the development of AAA.
More recently, Koole et al. 26 performed a study comparing concentration of three AGEs (pentosidine, carboxyethyllysine and CML) in aortic wall biopsies from AAA patients with and without DM as well as non-AAA patients with and without DM to investigate the role AGEs play in protecting diabetic patients against AAA development. They found increased pentosidine concentrations were associated with DM in aortic tissues from AAA (p = 0.02) and non-AAA (p = 0.02) patients. Among diabetic AAA patients, pentosidine concentrations negatively correlated with aortic diameter (r =−0.43, p = 0.02) and experimentally glycation of AAA tissue was associated with a significant reduction in MMP-induced, but not cathepsin-induced, proteolysis. No significant differences were observed for carboxyethyllysine or CML leading to the authors to conclude that cross-linking AGEs like pentosidine, but not non-cross-linking AGEs, play a role in protecting against AAA progression in diabetic patients. 26 To study the diverging effects of DM on AAA and peripheral arterial disease pathogenesis further, de Vos et al. 27 plan to perform a multicentre study that will measure the amount, type and location of AGEs in the arterial wall of AAA and peripheral arterial disease patients with and without DM, and then correlate these findings with pulse wave velocity measurements.
AGEs also maintain vessel wall architecture by rendering ECM proteins less susceptible to protein degradation and by influencing the production of cathepsins and MMPs. 28 Grzebyk et al. 29 found that circulating levels of AGEs were higher among patients with T2DM (p < 0.001) and that high AGE levels were associated with reduced plasma (p < 0.05) and neutrophil-derived (p < 0.01) levels of cathepsin B activity. Zhang et al.25,30 have also reported that AGEs can increase the production of MMP-9 in cultured macrophages but not in cultured AoSMCs. This pathway is regulated by extracellular signal-regulated kinases and nuclear factor kappa B (NF-κB) and can be attenuated by TGF-β1. 30 Golledge et al. 31 have shown that human monocytes exposed to glycated collagen lattices decrease their production of MMP-2 and MMP-9 which are important in AAA development. A similar phenomenon could be achieved by exposing monocytes to non-glycated but cross-linked collagen lattices which led the authors to suggest that it is the ability of hyperglycaemia to induce matrix protein cross-linking rather than hyperglycaemia itself that is responsible for the decrease in monocyte-derived MMP production. 31
Inflammation
The role of inflammation in explaining the negative relationship between DM and AAA is complicated. Up-regulation of inflammatory cell pathways is an essential component of AAA formation. 32 DM is known to induce vascular inflammation in atherosclerotic tissues;33,34 however, some studies also suggest that a similar phenomenon occurs among aneurysmal tissues which would seem inappropriate given the relationship between DM and AAA. 35 Indeed, Arapoglou et al. 35 examined macrophage levels in patients undergoing open AAA repair and found significantly higher levels in aortic tissues from patients with DM compared to those without (p = 0.02) and a positive correlation with serum glucose levels (r = 0.21, p = 0.04).
Others, however, have reported reduced vascular inflammation in DM. Miyama et al. 36 have shown, using the apolipoprotein E (ApoE)-deficient angiotensin II (AngII) and porcine pancreatic elastase mouse models of AAA, that hyperglycaemia (produced using streptozotocin administration) was associated with smaller AAA and a diminished inflammatory cell response characterised by a reduction in aortic macrophage infiltration (p = 0.03) and peritoneal macrophage production. In this study, insulin administration was associated with abrogation of AAA expansion. Hurks et al. 37 investigated the relationship between AAA inflammation and atherosclerotic risk factors and found that lymphocyte-poor (i.e. low levels of inflammation) rather than lymphocyte-rich aortic specimens were more commonly associated with many traditional atherosclerotic risk factors including DM (22% vs 9%; p = 0.008).
The exact mechanisms by which DM alters inflammation in AAA may involve the monocyte-macrophage system, 36 activation of T-cell insulin receptors 38 or through production of C-peptide. 39 Tanaka et al. 40 have recently shown that hyperglycaemia was able to suppress MMP-9 messenger RNA (mRNA; 17.9 ± 2.5 vs 50.5 ± 9.7; p < 0.01) and protein (3.6 ± 0.5 vs 6.4 ± 0.3; p < 0.01) expression in macrophage cell lines stimulated by tumour necrosis factor (TNF)-α and calcium phosphate. The authors went on to demonstrate that this suppression was mediated by hyperglycaemia-induced activation of the glucose-sensing nuclear receptor Nr1h2 (0.8 ± 0.4 vs 0.2 ± 0.04; p < 0.05). 40 Patients with type 2 diabetes mellitus (T2DM) often have raised circulating c-peptide levels. 41 Cifarelli et al. 42 have shown that physiological levels of c-peptide are capable of reducing hyperglycaemia-induced vascular smooth muscle cell proliferation, one of the primary lesions in diabetic atherosclerotic disease. Haidet et al. 43 have also shown that addition of c-peptide to monocytic cell lines exposed to high glucose was able to reduce expression of various pro-inflammatory cytokines via a NF-κB-mediated pathway.
ILT biology
The ILT has been implicated in the pathogenesis of AAA and indeed patients with a greater arc of thrombus covering the aneurysm wall have been shown to exhibit more rapid AAA expansion (p < 0.005). 44 It is believed that MMPs within the thrombus are released during thrombus renewal, a process that involves fibrinolysis, therefore thrombi that are more resistant to fibrinolysis may slow the rate of AAA expansion. 7 Dunn et al. 45 have shown that fibrin clots in patients with T2DM were denser, less porous and therefore less susceptible to fibrinolysis. Hyperinsulinaemia, a feature of T2DM, may also exert a protective effect on AAA by preventing renewal of the ILT. 46 By increasing PAI-1 levels, hyperinsulinaemia inhibits the conversion of plasminogen to plasmin which suppresses fibrinolysis 46 and also reduces MMP expression since plasmin is needed to convert pro-MMP to its active form.20,47
Oral hypoglycaemic agents
The three classes of oral hypoglycaemic agents that have received the most attention with respect to explaining the protective effect of DM on AAA are the biguanides (in particular metformin), the thiazolidinediones (TZDs) and dipeptidyl peptidase-4 (DPP4) inhibitors.
Thompson et al. analysed data on 1269 patients under AAA surveillance and were the first to report an association between medications used to treat DM and a reduction in AAA growth rates (56% reduction; p = 0.003 even after adjusting for confounders). The two classes of anti-diabetic drugs that were particularly associated with a reduction in AAA growth rates in this study were the biguanides (p = 0.05) and sulphonylureas (p = 0.03). 48 Fujimura et al. 49 performed a similar analysis using a much smaller cohort of patients but found that of the 11 classes of medications tested, only metformin demonstrated a significant negative association with AAA growth (p < 0.05). The authors went on to perform various mechanistic studies. They found that metformin administration to mice infused with porcine pancreatic elastase resulted in less AAA formation which was associated with medial elastin and SMC preservation, reduced B cell, macrophage, CD4 and CD8 T cell accumulation and reduced mural neovessel concentrations on histological studies. 49 Similar findings were demonstrated by Vasamsetti et al. 50 using the ApoE-deficient AngII mouse model. They found that metformin administration was associated with a significant reduction in maximal aortic diameter, incidence of aneurysm formation and aortic wall and MMP-1 and MMP-9 levels. Using a monocytic cell line they also showed that metformin reduced differentiation of monocytes into macrophages. 50
Hsu et al. 51 performed a contemporary analysis of Taiwan’s National Health Insurance database (>1.2 million patients) to identify 4468 patients with aortic aneurysms and 4468 matched controls. They found metformin, sulphonylurea and TZD use were associated with a significantly lower risk of developing aortic aneurysms (OR, 0.72, 0.82 and 0.82, respectively) and that the effects of metformin and sulphonylurea were dose-responsive (p < 0.001 for trend with both). No protective effect was found for α-glucosidase inhibitors or DPP4 inhibitors. 51 Most recently, Golledge et al. studied the effect of metformin, among other therapies, on AAA growth rates using three distinct patient cohorts. Prescription of metformin, but no other therapy for DM, was associated with a reduced likelihood of median or greater AAA growth in all three patient cohorts (OR range, 0.13–0.59; p < 0.02 in all cohorts). 52
Interestingly, Kristensen et al. 53 were concerned that metformin prescription might reverse the protective effect of DM on AAA formation and therefore examined the risk of ruptured AAA among individuals with and without DM using a nested case–control analysis of the national Danish registry data. They found that metformin prescriptions were not associated with an increased risk of ruptured AAA and may even protect against ruptured AAA although this did not reach statistical significance after correcting for confounders [OR, 0.84; 95% confidence interval (CI), 0.61–1.17]. 53
TZDs increase insulin sensitivity via activation of peroxisome proliferator-activated receptor gamma. Jones et al. have shown pre- or post-treatment with rosiglitazone in ApoE-deficient mice prevented Ang II-mediated aneurysm rupture and reduced maximal aortic diameter. Pre-treatment was also associated with a reduction in expression of the inflammatory mediators TNF-α and interleukin (IL)-6. 54 The same group went on to demonstrate that the actions of rosiglitazone on aneurysm formation were mediated via a reduction in c-jun N-terminal kinase expression throughout the aorto-iliac tree and toll-like receptor 4 expression selectively in the suprarenal aorta which represents the site of aneurysm formation in the Apo-E-deficient AngII model. 55 Using the same experimental mouse model, Golledge et al. 56 have produced similar reductions in aneurysm formation using another TZD pioglitazone (p = 0.01) and the related compound fenofibrate (p = 0.001) compared to using a vehicle control. Both compounds were also associated with reduced macrophage infiltration. Cockerill’s group suggested that pioglitazone may reduce experimental aneurysm formation via an action on the polycystic kidney disease 1 gene which itself is negatively controlled by early growth response protein-1. 57
The only study to test the effect of TZDs on AAA parameters in humans was performed by Motoki et al. This study found that pre-treatment of patients awaiting open AAA repair with 2 weeks of pioglitazone resulted in a significant reduction in macrophage infiltration, MMP-9 and TNF-α levels in both aortic wall and periaortic fat samples. 58
DPP4 inhibitors work by augmenting the incretin effect. Lu et al. 59 have shown a positive correlation between the relative intensity of plasma DPP4 and AAA diameter (p < 0.05) which may be mediated through an action of DPP4 on monocyte-macrophage differentiation. Using the DPP4 inhibitor alogliptin, Bao et al. were able to attenuate aneurysm formation in a dose-dependent fashion (p < 0.02) using rodent models of AAA (intraluminal elastase and extraluminal calcium chloride). Furthermore, they found that alogliptin treatment was associated with lower tissue levels of MMP-2 (p < 0.001), MMP-9 (p < 0.001) and reactive oxygen species (p < 0.0001) and preservation of mural elastin. 59 Kohashi et al. 60 performed a similar study using the DPP inhibitor MK0626 in an ApoE-deficient AngII mouse model and found that treatment with MK0626, but not treatment with native incretins GIP-1 or GLP-1, was associated with a 30% reduction in aneurysm formation, significantly less IL-1β expression and an increased ratio of tissue inhibitor of metalloproteinase (TIMP)-2 to MMP-9. MK0626 did not affect aortic IL-6 or TNF-α expression in this study.
Additional mechanisms
Studies that do not fall into any of the biological pathways detailed above but do provide evidence that may aid our understanding of the relationship between DM and AAA are listed here for completeness. A genetic basis for the relationship between DM and AAA seems unlikely following the results of a recent study using Mendelian Randomisation analyses 61 although post-genomic factors may be implicated. MicroRNAs are small non-coding RNA molecules that function in RNA silencing and regulating post-transcriptional gene expression. Maegdefessel et al. reported increased expression of miR-29b in experimental models of AAA. Suppression of miR-29b, which may occur with medications for DM, has been shown to promote a fibrotic response which may therefore limit aneurysm expansion.62,63 This is plausible since miR-29 is a key microRNA up-regulated by hyperglycaemia and has important roles in insulin-stimulated glucose metabolism. 64 However, there are no studies that have yet tested the effect of oral hypoglycaemic agents on miR-29 expression.
In addition to the increased arterial wall stiffness caused by cross-linking and calcification in diabetic patients, Madi et al. 65 have shown that DM alters the SMC phenotype to one which is more migratory and less proliferative which may also increase vessel stiffness. In their study, saphenous vein SMCs from T2DM patients exhibited a phenotype that promoted vinculin-positive focal adhesions with disrupted actin cytoskeletons and disorganised α-actin networks, 65 vinculin being an important focal adhesion protein coupling the ECM to the actin cytoskeleton. 7
Neovascularisation is one of the pathological hallmarks of AAA. 66 However, chronic hyperglycaemia results in microangiopathy and microvessel occlusion, which may explain why Miyama et al. 36 found reduced levels of adventitial neovascularisation in aortic aneurysm samples from hyperglycaemic compared with euglycaemic mice (12 ± 8 vs 20 ± 6 vessels per high-powered field; p = 0.02).
Finally, some authors have suggested a role for cell division autoantigen 1 (CDA1) in mediating the protective effect of DM on AAA. CDA1 has previously been shown to enhance TGF-β signalling in both renal and vascular cells and knockout of CDA1 in diabetic mice is associated with a reduction in renal expression of TGF-β1 and matrix accumulation. 67 Li et al. 68 found that ApoE knockout in diabetic mice did not lead to the formation of aneurysms but ApoE + CDA1 double knockout in diabetic mice led to the formation of aneurysms in 40% of animals. Furthermore, the authors observed not only reduced TGF-β signalling and expression of various collagens but also increased inflammatory cell infiltrate and expression of MMP-2, all histological changes observed in native AAA formation. 68
Conclusion
The negative relationship between DM and AAA is robust; however, the mechanisms underlying this relationship represent an important research question. This review has identified at least five biological pathways by which this relationship might be mediated (see Figure 1). Future research should aim to exploit these pathways to aid development of a pharmacological agent capable of slowing or halting aneurysm progression. Most importantly, any agent developed based on these pathways must undergo rigorous testing prior to widespread use to ensure it does not induce impaired glucose tolerance as this would certainly limit its clinical utility.
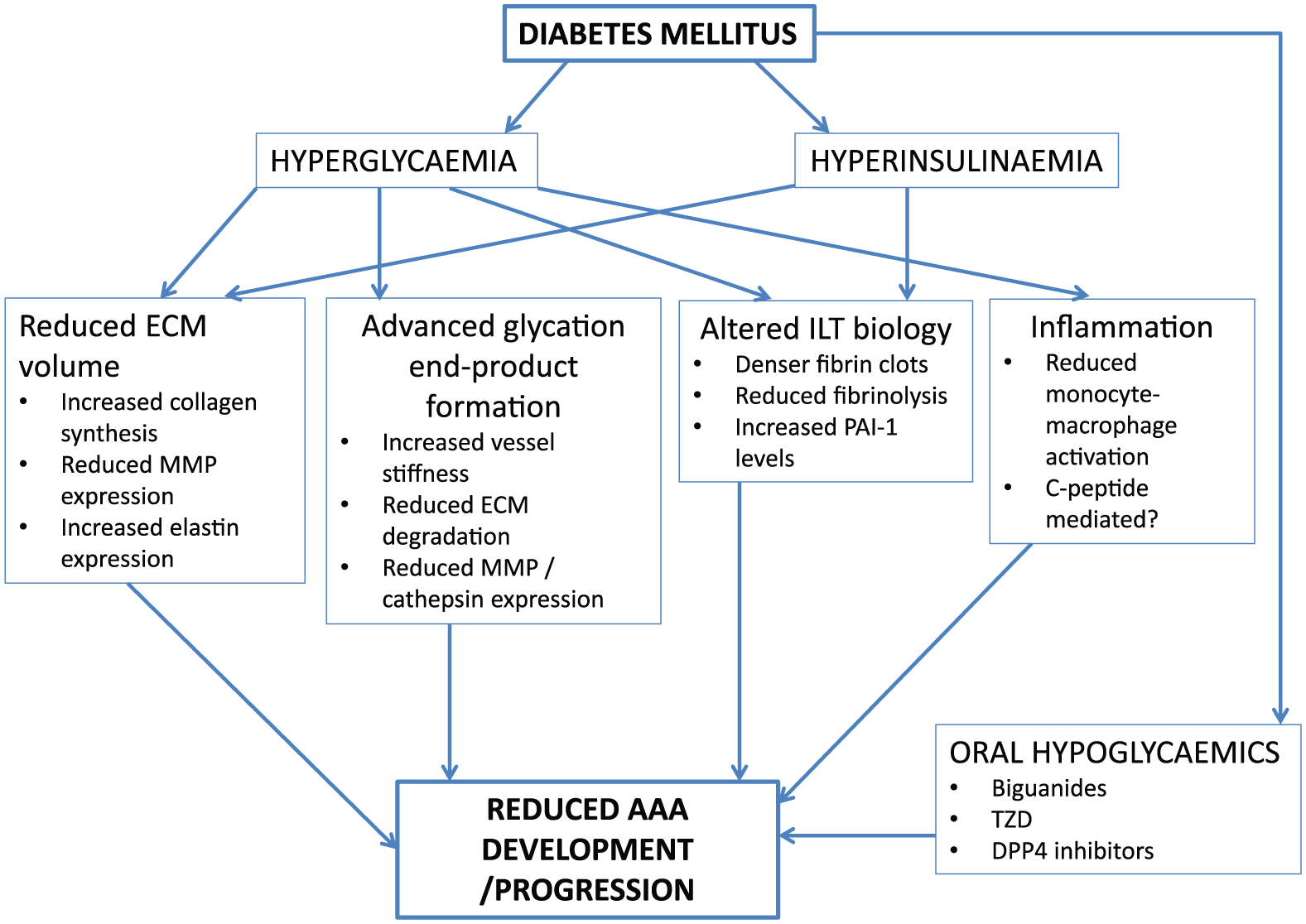
A summary of the potential mechanisms by which diabetes mellitus protects against abdominal aortic aneurysm development and progression. DPP4: dipeptidyl peptidase-4; ECM: extracellular matrix; ILT: intraluminal thrombus; MMP: matrix metalloproteinase; PAI: plasminogen activator inhibitor; TZD: thiazolidinedione.