
Abstract
Liraglutide, a once-daily glucagon-like peptide-1 receptor (GLP-1R) agonist, has been approved as a new treatment for type 2 diabetes and is the subject of a clinical trial programme to evaluate the effects on cardiovascular disease and safety. The current study aimed to determine the in vivo effect of liraglutide on progression of atherosclerotic vascular disease in the apolipoprotein E-deficient (ApoE−/−) mouse model and identify underlying mechanisms responsible. Liraglutide treatment inhibited progression of early onset, low-burden atherosclerotic disease in a partially GLP-1R-dependent manner in the ApoE−/− mouse model. In addition, liraglutide treatment inhibited progression of atherosclerotic plaque formation and enhanced plaque stability, again in a partially GLP-1R-dependent manner. No significant effect of liraglutide on progression of late onset, high-burden atherosclerotic disease was observed. In addition, no significant endothelial cell dysfunction was identified in ApoE−/− mice with early onset, low-burden atherosclerotic disease, although significant prevention of weight gain was observed in liraglutide-treated mice using this dietary protocol. Taken together, these results suggest a potential role for liraglutide in the prevention and stabilisation of atherosclerotic vascular disease together with possible protection against major cardiovascular events.
Keywords
Introduction
The incretin glucagon-like peptide-1 (GLP-1) has a well-documented role in glucose-stimulated insulin release and the preservation of plasma glucose homeostasis. 1 The importance of the incretin system in glucose homeostasis is emphasised by the recent successful therapeutic exploitation of this system utilising glucagon-like peptide-1 receptor (GLP-1R) agonists and inhibitors of the dipeptidyl peptidase 4 (DPP-4) enzyme in the treatment of type 2 diabetes. 2
Identification of the GLP-1R in tissues outside the pancreatic β-cell and in particular on vascular endothelial cells 3 suggests a role for GLP-1 in vascular biology. In vitro native GLP-1 and GLP-1R agonists attenuate tumour necrosis factor (TNF)-induced plasminogen activator inhibitor-1 (PAI-1) and vascular adhesion molecule expression in human vascular endothelial cells. 4,5 In vivo studies demonstrate that native GLP-1 and GLP-1R agonists regulate vascular tone and endothelial function, 6 –8 and clinical studies have reported reduced endothelial dysfunction in type 2 diabetic patients with established coronary artery disease. 3 Several recent in vivo studies identify atheroprotection by native GLP-1, GLP-1R agonists and DPP-4 inhibitors. 9 –12 Given type 2 diabetes is characterised by accelerated atherosclerosis, 13 the potential atheroprotective effects of GLP-1, long-acting GLP-1R agonists and DPP-4 inhibitors are currently the subject of intense preclinical and clinical researches.
Liraglutide, an acylated GLP-1R agonist, has a half-life of 10–14 h allowing once-daily subcutaneous (s.c.) injection and has been approved as a new treatment for type 2 diabetes. 14 Clinical trials of liraglutide in patients with type 2 diabetes demonstrate improved levels of both fasting and postprandial glucose, improvement in β-cell function, a reduction in plasma glucagon concentration, weight loss, reduced blood pressure and an improvement in cardiovascular risk biomarkers. 14 –17 Recent in vivo studies indicate that liraglutide may also influence the progression of prediabetes to diabetes. 18
Liraglutide attenuates endothelial cell dysfunction, a known predictor of cardiovascular events, together with in vivo expression of vascular adhesion molecules, and is the subject of a clinical trial programme to evaluate efficacy in cardiovascular disease prevention. 8,19 The current study aimed to determine the in vivo effect of liraglutide on atherosclerotic vascular disease in the apolipoprotein E-deficient (ApoE−/−) mouse model at different stages of disease progression and identify underlying mechanisms responsible.
Materials and methods
Materials
Liraglutide was provided by Novo Nordisk A/S, Denmark. Exendin-9 was purchased from GL Biochem (Shanghai) Ltd, Shanghai, China.
In vivo studies
Non-diabetic male B6 ApoE−/− mice with a >99% C57BL/6J background were obtained from Animal Resources Centre (Canning Vale, WA, Australia). All mice were obtained at 5 weeks of age and weighed between 25 and 35 g at the time of experiments. Animal experiments were performed in accordance with the Guide for the Care and Use of Laboratory Animals published by the US National Institute of Health, and these were approved by the Monash University School of Biomedical Sciences Animal Ethics Committee.
Blood pressure measurement
Systolic blood pressure (SBP) was measured prior to and at the end of treatment using non-invasive tail-cuff plethysmography (AD Instruments, Sydney, NSW, Australia).
Treatment protocols
In vivo experiments utilised either (1) early onset, low-burden disease protocol or (2) established, high-burden disease protocol: (1) 17-week-old ApoE−/− mice maintained on a normal chow diet were simultaneously commenced on a high-fat diet (HFD) containing 22% fat and 0.15% cholesterol (Specialty Feeds, Glen Forrest, WA, Australia) and saline (vehicle: n = 15), liraglutide (300 µg/kg twice daily, s.c.; n = 17) or liraglutide + exendin-9 (a specific GLP-1R antagonist; 150 pmol/kg/min s.c., via osmotic mini-pump, n = 13) for 4 weeks or (2) 18-week-old ApoE−/− mice maintained on a HFD for 12 weeks were treated with a regimen identical to protocol 1 for 4 weeks – saline (vehicle: n = 14), liraglutide (n = 10) and liraglutide + exendin-9 (n = 6). Implantation of the mini-osmotic pumps occurred under general anaesthetic with ApoE−/− mice anaesthetised using inhaled isoflurane and an incision made in the midscapular region through which mini-osmotic pumps (ALZET model 1004; Alza Corp., Cupertino, CA, USA) were inserted for subcutaneous drug administration. Dosage of exendin-9 was selected based on previous in vivo study results. 20 At the end of treatment, animals were anaesthetised by isoflurane inhalation and decapitated. Heart and the whole aorta were removed and placed in ice-cold Krebs bicarbonate buffer (pH 7.4) consisting of 118 mM NaCl, 4.7 mM KCl, 1.2 mM KH2PO4, 1.2 mM MgSO4·7H20, 2.5 mM CaCl2, 25 mM NaHCO3 and 11.7 mM glucose. The connective tissue surrounding the aorta was carefully removed under a binocular dissection light microscope (Olympus SZ40; Olympus, Shinjuku, Tokoyo, Japan), and the abdominal aorta was dissected for vascular reactivity studies, leaving the aortic arch and thoracic aorta attached to the heart for further histological analysis.
Histological analysis of samples
Oil red O staining (protocol 1, n = 13–16; protocol 2, n = 6–14) and intima:media ratio (IMR) analysis (protocol 1, n = 6–10; protocol 2, n = 5–10) were performed in the aorta and aortic arch, respectively. Plaque stability was analysed in the brachiocephalic artery (BCA), a well-recognised model for human atherosclerotic plaque formation and rupture being subjected to high shear rates and developing plaques at a similar rate and with similar morphology to those identified in humans. 21 Plaque stability was measured via analysis of vascular smooth muscle cell α-actin content, lipid deposition, collagen content and macrophage staining within plaques using cross sections of this artery (protocol 1, n = 5–7). Specifically, cryocut cross sections (5 µm) of the BCA were prepared, and two sections (20 µm apart) for each mouse were stained routinely with (1) oil red O (0.5% saturated oil red O) for lipid deposition and imaged under a normal bright-field light, (2) with picrosirius red (0.05% saturated picrosirius red) for collagen content and imaged under a polarised light or (3) immunostained with either a primary rat monoclonal antibody specific for monocytes/macrophages (F4/80; Serotec, Raleigh, NC, USA) or a primary rabbit polyclonal α-smooth muscle actin (α-SMA) antibody (Dako, Glostrup, Denmark), then visualised by incubation with either a polyclonal goat anti-rat IgG conjugated with Alexa488 (Invitrogen, Carlsbad, CA, USA) or goat anti-rabbit IgG conjugated with Alexa594 (Invitrogen) secondary antibody. Immunofluorescent sections were viewed under 20× magnification (BX51, Olympus), and images were taken. The percentage of immunofluorescence in the atherosclerotic plaque was quantified using ImageJ 1.46 software (Java, NIH, Bethesda, MD, USA) and averaged out from the two sections per animal. Plaque stability score was calculated via both the collagen area-to-lipid area ratio and the formula (collagen + smooth muscle cell area)/(macrophage + extracellular lipid deposition area) in the plaque of the BCA. 22,23
Vascular reactivity studies
Four aortic rings per mouse (~3 mm long) were obtained from the abdominal aorta, and each ring was suspended between two stainless steel wires connected to an isometric force transducer (FT-03; Grass Instruments, Warwick, RI, USA) (protocol 1, n = 7–9). Concentration–response curves to the endothelium-dependent vasodilator acetylcholine (ACh) were constructed in tissues preconstricted with the thromboxane A2 analogue, U46619 ([1,5,5-hydroxy-11α, 9α-(epoxymethano)prosta-5Z, 13E-dienoic acid]), as previously described. 24 At the end of the ACh curve, 10 µM of the endothelium-independent vasodilator, sodium nitroprusside (SNP), was added to the bath to test vascular smooth muscle integrity.
Data analysis
All results are expressed as mean ± standard error of the mean (SEM). Statistical comparisons were by one-way or two-way repeated measures analysis of variance (ANOVA) with the Bonferroni corrections where appropriate using GraphPad Prism version 4.00 (GraphPad Software, San Diego, CA, USA). The non-parametric statistical Kruskal–Wallis test was used for immunohistochemical analysis. A value of p < 0.05 was deemed statistically significant.
Results
Liraglutide attenuates progression of early onset, low-burden atherosclerotic disease
Oil red O staining and IMR analysis of atherosclerotic plaques from the aorta identified attenuation of lipid deposition (liraglutide group, 5.99% ± 0.96%, vs vehicle group, 10.02% ± 1.34%) and a significant reduction of IMR in liraglutide-treated mice (liraglutide group, 0.11 ± 0.03, vs vehicle group, 0.30 ± 0.07, p < 0.05) from early onset, low-burden atherosclerotic disease associated with protocol 1 (Figure 1(a) to (c)). This effect was, in-part, attenuated by concomitant addition of exendin-9, suggesting a GLP-1R-mediated effect. No significant effect on inhibition of oil red O staining or IMR of liraglutide treatment on mice from protocol 2 was identified, although a trend towards a reduction in IMR was observed, suggesting a possible effect on atherosclerotic disease progression in the setting of established or high-burden atherosclerotic disease (Figure 2(a) to (c)).
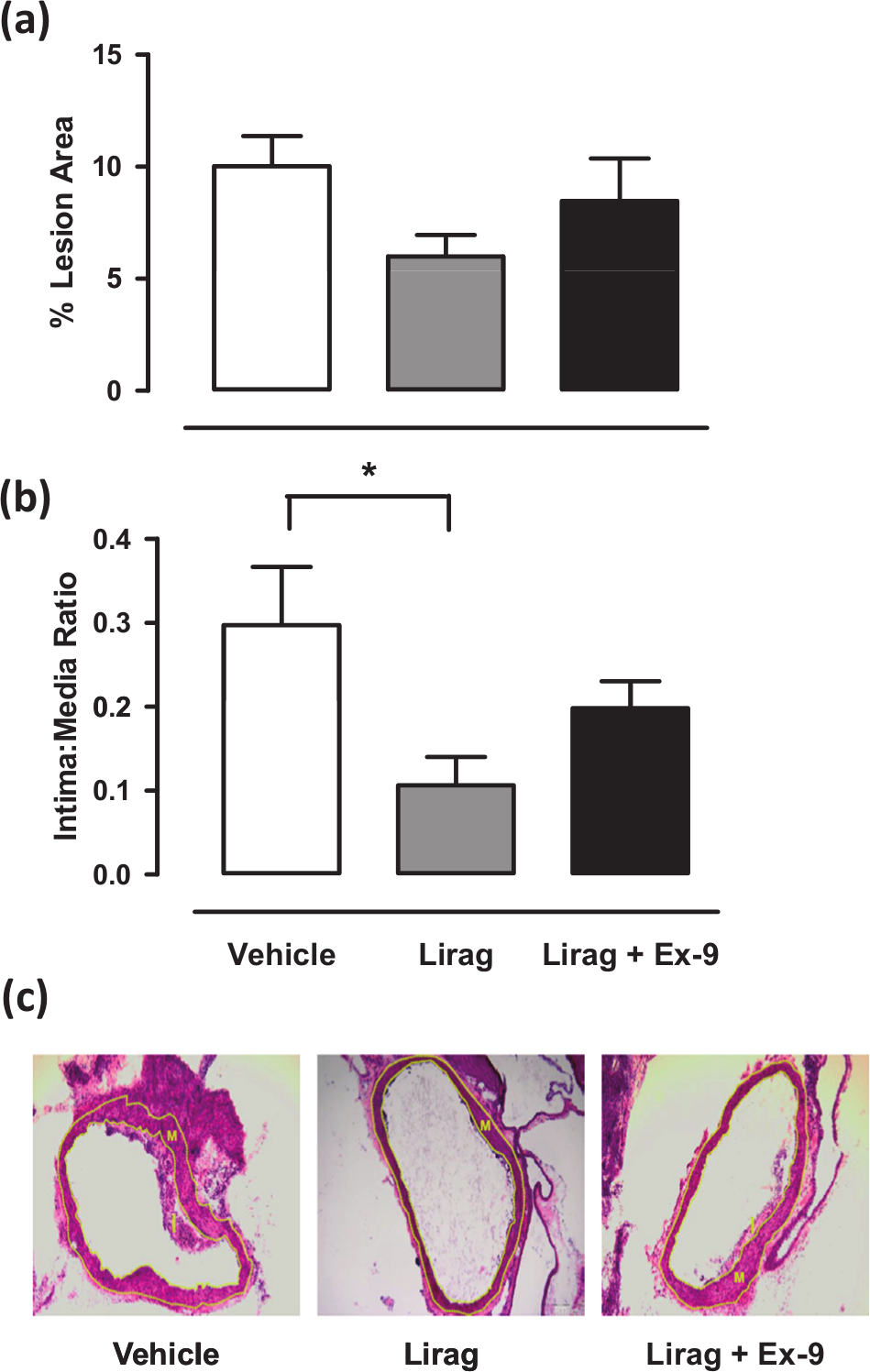
Effect of liraglutide treatment on early, low-burden atherosclerotic lesion development in ApoE−/− mice: (a) Mean ± SEM data for percentage lesion area measured using oil red O staining from all treatment groups in protocol 1 (n = 13–16), (b) mean ± SEM data for IMRs from all treatment groups from protocol 1 (n = 6–10) (*p < 0.05 vs vehicle, one-way ANOVA) and (c) representative photographs (stained with haematoxylin and eosin) of cross-sectional lesion development in the aortic arch; intima and media areas (border identified with dotted yellow line) were measured using the AnalySIS program and represented as IMR (20× magnification).
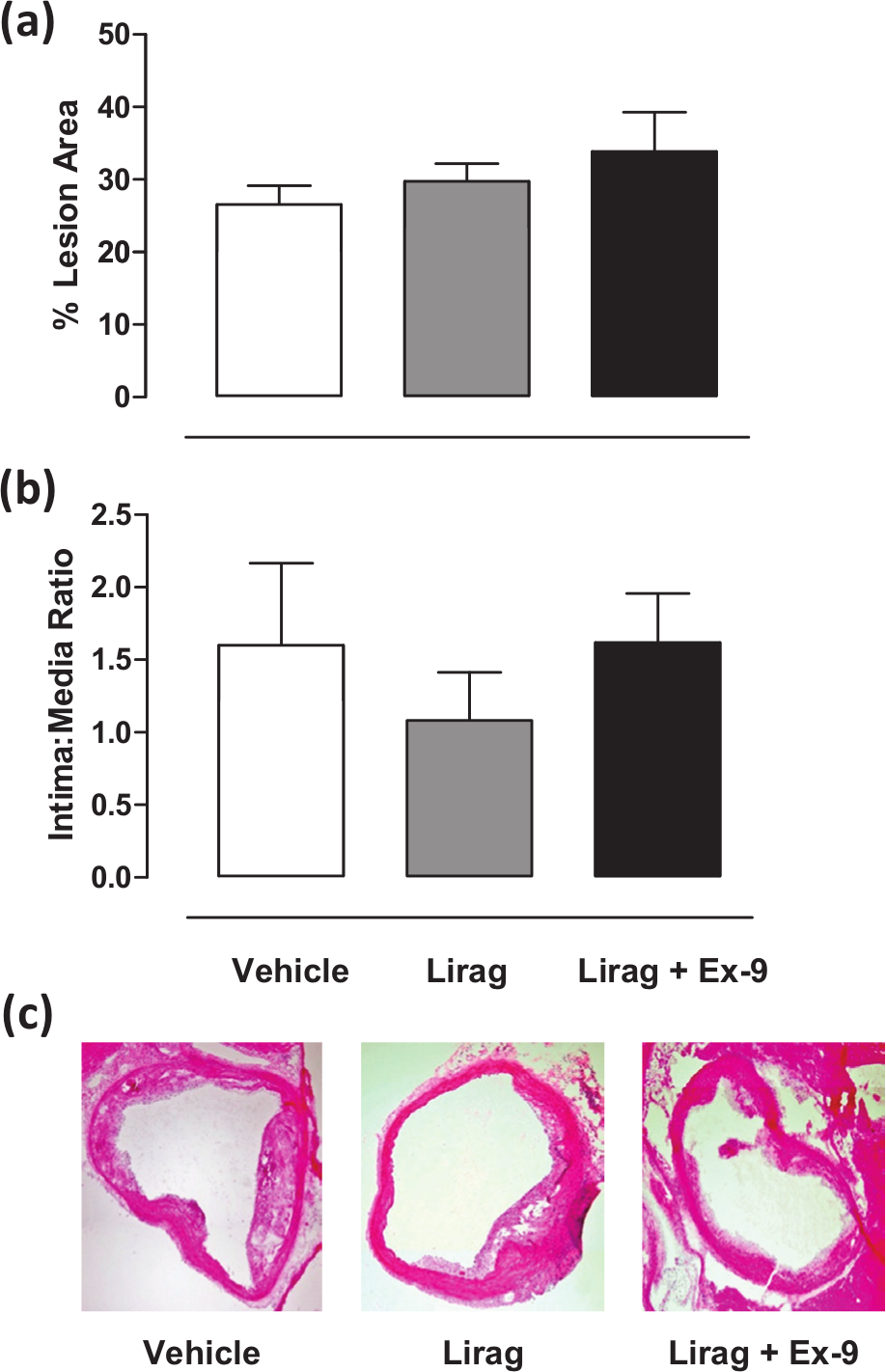
Effect of liraglutide treatment on established, high-burden atherosclerotic lesion development in ApoE−/− mice: (a) Mean ± SEM data for percentage lesion area measured using oil red O staining from all treatment groups in protocol 2 (n = 6–14), (b) mean ± SEM data for IMRs from all treatment groups from protocol 2 (n = 5–10) and (c) representative photographs (stained with haematoxylin and eosin) of cross-sectional lesion development in the aortic arch; intima and media areas were measured using the AnalySIS program and represented as IMR (20× magnification).
Liraglutide significantly enhances the stabilised plaque phenotype in early onset, low-burden atherosclerotic disease
To investigate the potential effects of liraglutide on atherosclerotic plaque stability, we utilised the BCA from ApoE−/− mice on dietary protocol 1. A significant decrease in lipid deposition within atherosclerotic plaques assessed [liraglutide: 30.50% ± 9.84% vs vehicle: 61.86% ± 8.06%, p < 0.05 (Figure 3(a))] together with a trend to increase in collagen content [liraglutide: 24.08% ± 8.38% vs vehicle: 8.69% ± 3.07% (Figure 3(b))] and a significant increase in vascular smooth muscle cell content [liraglutide: 36.53% ± 8.79% vs vehicle: 12.84% ± 4.46%, p < 0.05 (Figure 3(c))] within BCA atherosclerotic lesions in liraglutide-treated mice from protocol 1 was identified. These effects were, in-part, attenuated by concomitant addition of exendin-9, suggesting a GLP-1R-mediated effect. No significant effects on macrophage deposition were observed (Figure 3(d)).
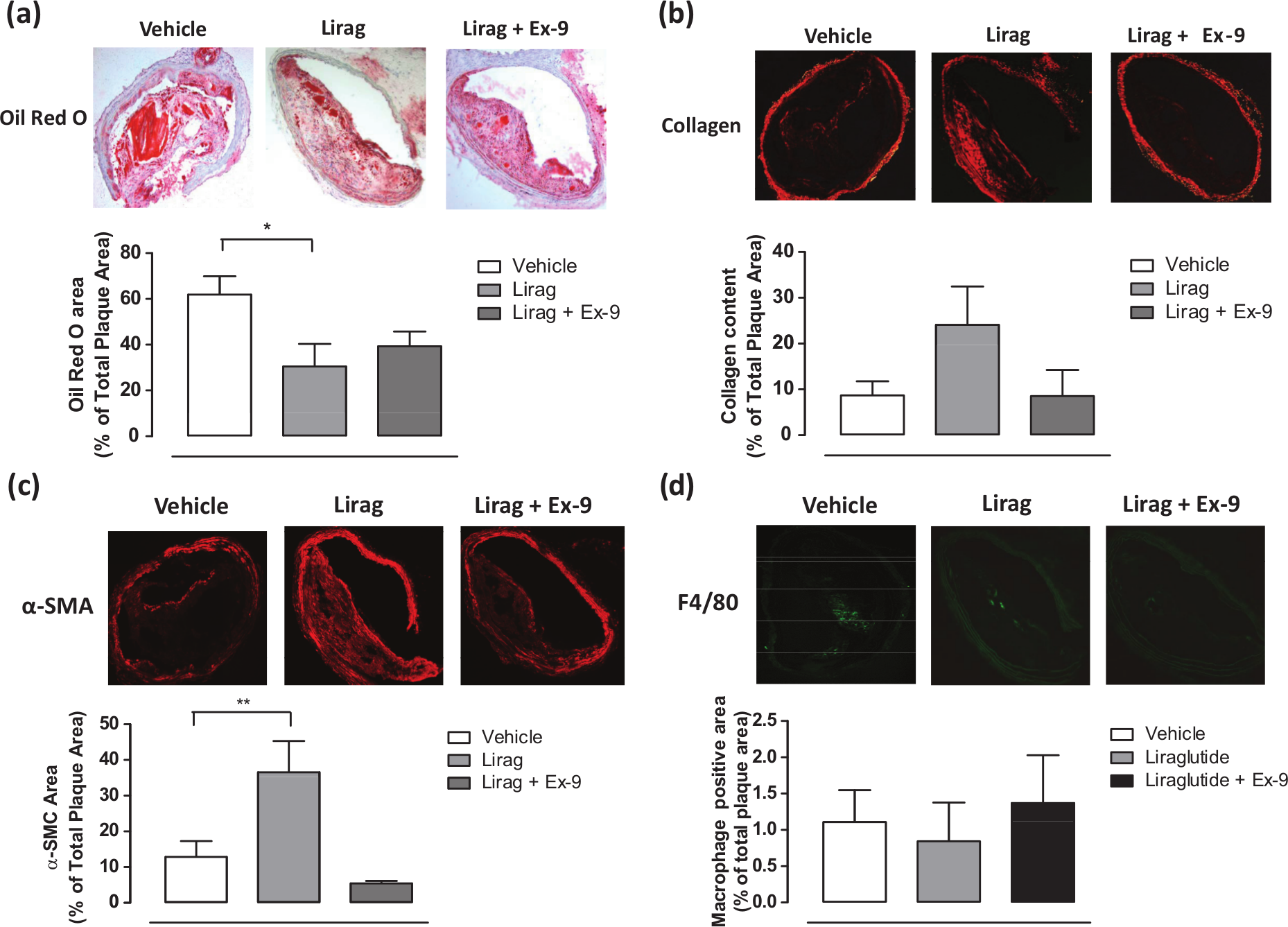
Liraglutide treatment significantly improves plaque stability in early, low-burden atherosclerotic disease in ApoE−/− mice: top panel: representative images of brachiocephalic artery cross sections (20× magnification) and bottom panel: mean data of (a) lipid, (b) collagen deposition, (c) VSMC and (d) macrophage content in brachiocephalic artery atherosclerotic plaques of vehicle, liraglutide and liraglutide + exendin-9 treated ApoE−/− mice from protocol 1.
Liraglutide significantly increases plaque stability score in the BCA of ApoE−/− mice with early onset, low-burden atherosclerotic disease
The plaque-stabilising effect associated with liraglutide treatment as assessed by plaque stability score determined by plaque collagen content divided by oil red O staining area demonstrated a significant, in-part GLP-1R-dependent, improvement in BCA plaque stability from liraglutide-treated ApoE−/− mice with early onset, low-burden atherosclerotic disease [vehicle: 0.13 ± 0.07; liraglutide: 0.50 ± 0.11; liraglutide + exendin-9: 0.22 ± 0.14 (Figure 4)]. Calculation of plaque stability score using the following formula also demonstrated a significant improvement in BCA plaque stability (data not shown): (collagen + smooth muscle cell area)/(macrophage + extracellular lipid deposition area).
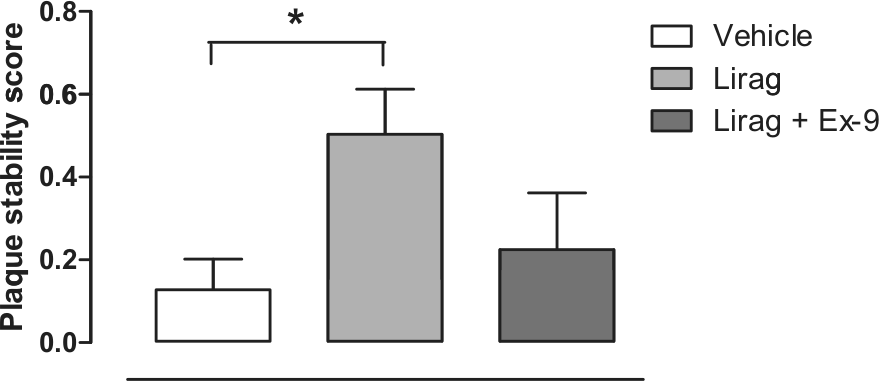
Liraglutide treatment significantly improves plaque stability score in early, low-burden atherosclerotic disease in ApoE−/− mice. Plaque stability calculated as described in methods.
Liraglutide attenuates endothelial cell dysfunction in established, high-burden, but not in early onset, low-burden, atherosclerotic disease
No significant endothelial dysfunction was observed in mice from dietary protocol 1 with liraglutide treatment resulting in no reduction in the level of vasorelaxation induced by ACh in these animals (Figure 5).
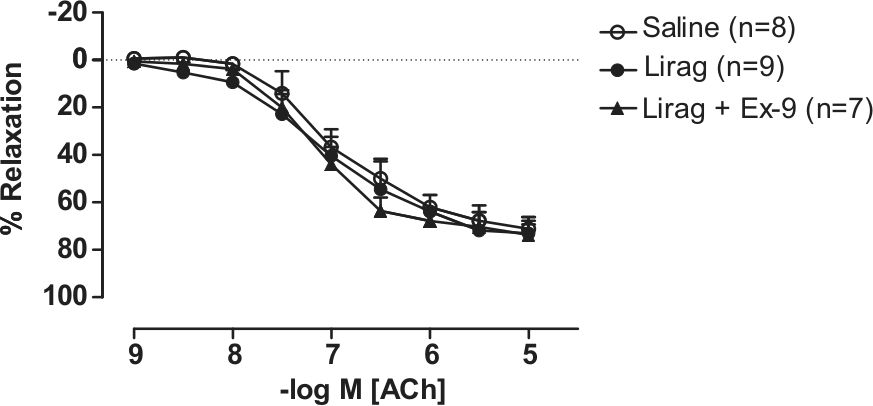
Effect of liraglutide treatment on endothelial function in aorta of ApoE−/− mice with early onset, low-burden atherosclerotic disease. Concentration–response curve to the endothelium-dependent vasodilator ACh in abdominal aorta taken from ApoE−/− mice from protocol 1 that received vehicle (saline), liraglutide [300 µg/kg (80 nmol/kg) twice daily, s.c.] or liraglutide + exendin-9 (150 pmol/kg/min) for 4 weeks, n = 7–9.
Liraglutide attenuates weight gain and increase in SBP in early onset, low-burden atherosclerotic disease
A significant increase in body weight was observed in ApoE−/− mice after administration of a HFD for 4 weeks (31.14 ± 0.59 g vs 33.77 ± 0.75 g, p < 0.01). No significant increase in body weight was observed in protocol 1 liraglutide-treated ApoE−/− mice after commencing a HFD (30.35 ± 0.52 g vs 29.96 ± 0.42 g) (Figure 6). A trend towards an increase in SBP was observed in ApoE−/− mice after administration of a HFD for 4 weeks. No increase in SBP was observed in liraglutide-treated ApoE−/− mice after commencing a HFD (Table 1).
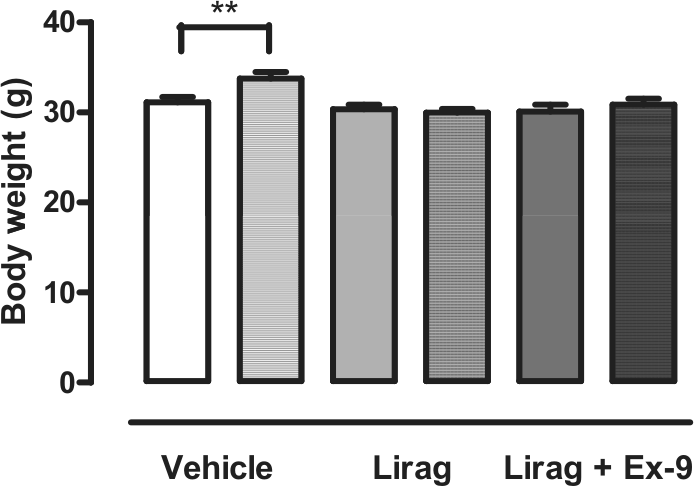
Effect of liraglutide treatment on body weight in ApoE−/− mice with early onset, low-burden atherosclerotic disease. Body weight (g) from vehicle (saline), liraglutide [300 µg/kg (80 nmol/kg) twice daily, s.c.] or liraglutide + exendin-9 (150 pmol/kg per min) treated mice from protocol 1 before and after (hatched bars) commencement of high-fat diet.
Effect of liraglutide treatment on SBP (mmHg) in ApoE−/− mice.
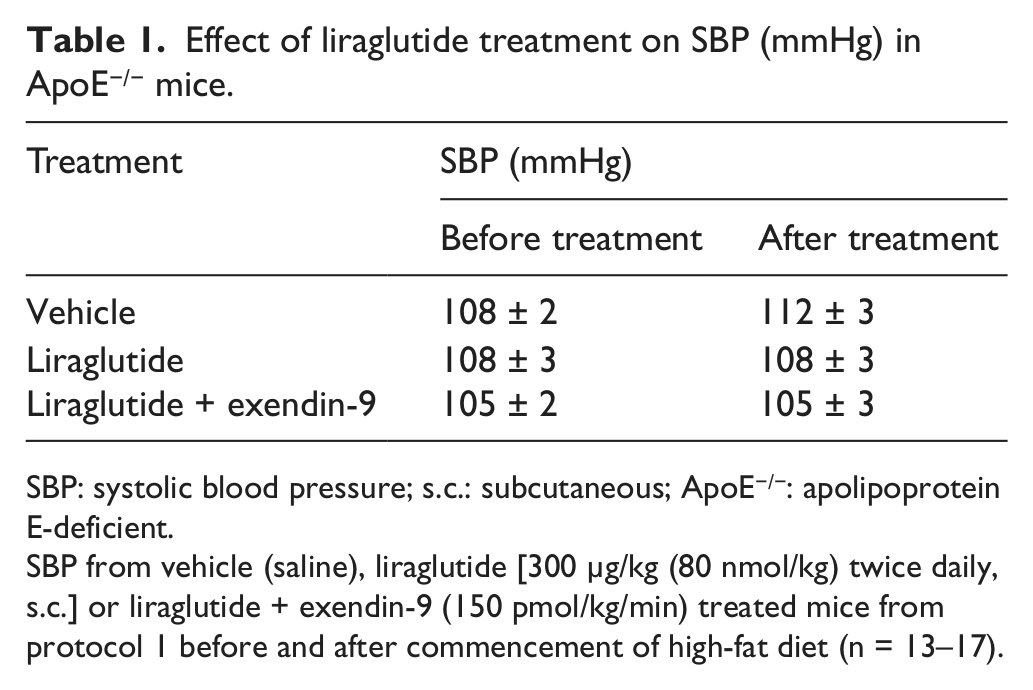
SBP: systolic blood pressure; s.c.: subcutaneous; ApoE−/−: apolipoprotein E-deficient.
SBP from vehicle (saline), liraglutide [300 µg/kg (80 nmol/kg) twice daily, s.c.] or liraglutide + exendin-9 (150 pmol/kg/min) treated mice from protocol 1 before and after commencement of high-fat diet (n = 13–17).
Discussion
Recent in vivo studies suggest that incretin-based therapies, in addition to improvement in glycaemic control, may afford protection from initiation and progression of atherosclerotic vascular disease via anti-inflammatory mechanisms together with inhibition of monocyte adhesion and macrophage infiltration into the vessel wall. 9 –11,25 These observations together with additional in vivo studies and clinical findings identifying improvement in endothelial cell dysfunction and a reduction in body weight and SBP with native GLP-1 and GLP-1R agonists 3,8,17,18 suggest multiple mechanisms of potential vascular protection within the vascular disease continuum from lesion initiation to pathological cardiovascular event.
Our previous studies have identified GLP-1 and the GLP-1R agonist liraglutide as able to mediate inhibition of cytokine and mild hyperglycaemia–induced expression of vascular peptides in human vascular endothelial cells in vitro together with improvement in endothelial cell dysfunction and inhibition of vascular adhesion molecule expression in the in vivo ApoE−/− mouse model. 4,5,8 Furthermore, we have previously observed a small but significant reduction in body weight together with a trend towards reduction in SBP from liraglutide-treated ApoE−/− mice with established, high-burden atherosclerotic disease identified in protocol 2. 8 We were subsequently interested in determining the in vivo effect of liraglutide on progression of atherosclerotic vascular disease and importantly atherosclerotic plaque stability in the assessment of this GLP-1R agonist.
Our observations suggest liraglutide-mediated, in-part GLP-1R-dependent, inhibition of atherogenesis in the setting of low-burden, early onset atherosclerotic disease together with improvement in plaque stability, a previously undocumented observation for the GLP-1R agonist therapeutic class. Endothelial dysfunction is a frequently reported observation with respect to early disease models; however, we were unable to demonstrate any evidence of this occurring in the setting of low-burden disease, which may reflect a limited sensitivity of this assay rather than absolute absence of mild endothelial dysfunction. 26
In the setting of established, high-burden atherosclerotic disease, while a trend to attenuation in IMR was observed, no significant reduction in atherosclerotic disease was identified; however, this particular protocol has been previously associated with an improvement in endothelial dysfunction. 8 The capacity of incretin-modulating agents to influence plaque stability is not unprecedented with recent observations of increased plaque stability in the absence of a reduction in atherosclerotic disease burden being identified in DPP-4 inhibitor–treated ApoE−/− mice. 27 Interestingly, this study utilised a dietary protocol similar to protocol 2 used in our studies and suggests that plaque stabilisation can be initiated in early stage disease and continues with the establishment of high-burden atherosclerotic disease even in the absence of obvious treatment-mediated reduction in disease burden.
Additional recent studies have brought into question several components of ApoE−/− mouse experimental models of potential GLP-1R-mediated modulation of atherogenesis including animal age, duration of treatment and induction of hyperglycaemia. 28 These variables together with potential tissue heterogeneity in GLP-1R expression and hepatic lipid metabolism in this model may explain differences in anti-atherogenic responses to GLP-1R agonists observed. 28
While enhancement of plaque stability is associated with a demonstrable reduction in cardiovascular events, 29 biological-based agents used in the treatment of diabetic hyperglycaemia have, outside of the possible effects of insulin, 30 yielded few if any useful benefits with respect to enhancement of atherosclerotic plaque stability. Our observations in relation to plaque stability suggest a powerful in-part GLP-1R-dependent plaque-stabilising effect of liraglutide with demonstrable effects on numerous mechanistic components of the stabilised plaque phenotype including modulation of vascular smooth muscle cell numbers and collagen content. 29 The effects on plaque stability may also have contributions from the identified reductions in blood pressure and weight gain afforded by liraglutide.
Our observations provide for the first time a possible temporal scenario for the potential vascular benefits of treatment with liraglutide in the ApoE−/− mouse model. In the setting of early vascular disease, protection from ongoing atherogenesis is afforded through reduced plaque progression together with stabilisation of existing atherosclerotic plaques. With ongoing chronic stimulation of atherogenesis resulting in a higher disease burden, a limited capacity of treatment to attenuate atherosclerotic disease formation is observed, consistent with recent findings, although plaque stabilisation is likely to be ongoing during this period. 27 With more established high-burden disease, treatment-associated improvement in endothelial cell dysfunction together with durable improvements in SBP and attenuation of weight gain 8 are likely to combine to contribute to a phenotype with significant protection against future cardiovascular events.
Footnotes
Funding
This research was supported in part by Novo Nordisk A/S of Bagsværd, Denmark.