
Abstract
Glycation of low-density lipoprotein (LDL) increases its atherogenicity, but whether high-density lipoprotein (HDL) can protect LDL against glycation is not known. LDL and HDL were isolated from 32 volunteers with serum HDL cholesterol concentrations ranging from 0.76 to 2.01 (mean = 1.36) mmol/L. Glycation of LDL was induced by incubation with 0–80 mmol/L glucose for 7 days at 37°C under nitrogen in the presence of and absence of human HDL. Glycation of LDL apolipoprotein B (apoB) doubled at glucose 50 and 80 mmol/L (both p < 0.001), and this increase was ameliorated by HDL. In the absence of glucose, 0.11 (0.01) [mean (standard error, SE)] mg apoB/mg LDL protein was glycated increasing to 0.22 (0.02) mg/mg at glucose 80 mmol/L in the absence of HDL, but remaining at 0.13 (0.01) mg/mg when autologous HDL was present. Heterologous HDL from a further study of 12 healthy participants was similarly effective in impeding LDL apoB glycation. HDL impeded not only glycation but also the lipid peroxidation, free amino group consumption and increased electrophoretic mobility of LDL which accompanied glycation. HDL from participants with higher serum paraoxonase1 (PON1) was more effective in impeding glycation and the related processes. In conclusion, HDL can impede the glucose-induced glycoxidation of LDL. PON1 may be important for this function of HDL.
Introduction
Epidemiological studies have demonstrated that the incidence of atherosclerotic cardiovascular diseases correlates positively with low-density lipoprotein (LDL) and negatively with high-density lipoprotein (HDL). 1 For LDL to participate in processes leading to atherosclerosis, such as foam cell formation resulting from its uptake by monocyte/macrophages, it must be chemically modified. Modification of LDL by oxidation has attracted most attention in this context.2,3 HDL is effective in preventing in vitro oxidative modification of LDL both by transition metal ions4–6 and by cells in tissue culture. 7 HDL is associated with lower levels of conjugated dienes in plasma from healthy people and those with coronary heart disease (CHD), and human HDL infused into hypercholesterolaemic rabbits also exhibits the capacity to lower the plasma levels of conjugated dienes.8,9
Although there has been a great focus of attention on oxidation as the atherogenic modification of LDL necessary for atherogenesis, glycation of LDL has also been shown to permit monocyte/macrophage uptake of LDL by similar receptor-mediated pathways. 10 Even in non-diabetic, normolipidemic people, glycatively modified LDL concentrations are typically 2–3 mg/dL in terms of apolipoprotein B (apoB), and levels are substantially higher in hyperlipidaemia and in diabetes.10,11 LDL glycation might ultimately prove at least as important in atherogenesis as oxidation. We were, therefore, interested to know whether HDL showed a protective effect against in vitro non-enzymatic glycation of LDL. This interest was further stimulated by emerging evidence from our group and others that components of HDL may protect against microvascular complications of diabetes such as retinopathy and nephropathy12–22 in which glycation is believed to play a part. 22
Some 100 different proteins have been identified in HDL. 23 HDL composition in people who are atherosclerosis prone, have diabetes or are in a chronic inflammatory state is known to differ considerably from that of healthy people.23–25 Variations in its composition, particularly in paraoxonase1 (PON1), lecithin-cholesterol acyltransferase, phospholipase A2, serum amyloid A and apolipoprotein J have been linked with HDL dysfunction.5,23–25 Of these, low activity of PON1, an enzyme which is almost exclusively located on HDL, has been associated with a diminished capacity of HDL to protect LDL against oxidative modification and with accelerated atherosclerosis5,6,9,26–28 on the one hand and with diabetic microvascular complications12,14,17,19,20 on the other. While there is incomplete evidence that PON1 is itself directly protective against atherogenesis or microvascular disease, low serum PON1 activity may be regarded as a marker of HDL with a composition that renders it dysfunctional in these respects. The current study aimed to determine the capacity of HDL to protect LDL against glycation and oxidative modification induced in vitro by glucose and to establish whether PON1-rich HDL behaved differently.
Subjects and methods
Subjects
The 44 participants were patients who were attending the Lipid Clinic at Manchester Royal Infirmary (n = 20) and healthy volunteers working in Manchester University and Manchester Royal Infirmary (n = 24). From the Lipid Clinic 2 patients were diabetic according to the World Health Organization (WHO) criteria and 7 patients including one of those with diabetes received statin treatment. Blood was obtained by venepuncture, after an overnight fast, into BD-vacutainer® serum tubes (Nu-care products Ltd, Bedfordshire, UK). Serum was isolated by centrifugation at 2000 × g at 4°C for 15 min using a Heraeus Labofuge 400R centrifuge (DJB Labcare, Buckinghamshire, UK). All participants gave informed consent to the study, which was approved by the Local Research Ethics Committee.
Lipoprotein isolation and in vitro glycation
LDL (D = 1.019–1.063 g/mL) and HDL (D = 1.063–1.21 g/mL) were isolated from serum by sequential density gradient ultracentrifugation without the addition of ethylenediaminetetraacetic acid (EDTA) 29 in a Beckman L7 ultracentrifuge with a 50.4 Ti fixed angle rotor using 13 × 64 mm polyallomer tubes at speeds of 34,000 r/min (144,361 × g) for 22 h 17 min. The isolated fractions were dialysed against phosphate buffered saline (PBS) overnight at 4°C followed by sterile filtration (0.2 µm) and stored at 4°C under nitrogen for not more than 16 h before glycation studies. Isolated LDL (0.5 mg protein/mL) was incubated under nitrogen in airtight screw-capped 2 mL-polypropylene sample tubes with 0, 30, 50 and 80 mmol/L glucose in PBS containing 0.01% chloramphenicol and Ca2+ 2 mmol/L for 7 days at 37°C in the presence of and absence of autologous HDL (0.5 mg protein/mL in PBS with Ca2+ 2 mmol/L) in a Gallenkamp Economy Size 1 Incubator (Gallenkamp, Leicester, UK). In 12 further experiments, LDL from healthy volunteers was glycated in vitro with glucose (80 mmol/L) in the presence of their own HDL and with HDL from another participant (cross-over experiments). Following incubation, EDTA (5 mmol/L) and butylated hydroxytoluene (BHT; 20 µmol/L) were added to terminate lipid peroxide (LPO) formation and inhibit PON1 activity. Following dialysis overnight at 4°C against PBS to remove non-protein-bound glucose, aliquots were used for assessment of in vitro induced glycation of LDL, LPO concentrations, determination of free amino groups and relative electrophoretic mobility (REM).
Laboratory analyses
Glycated lipoproteins were separated from non-glycated ones using m
LPO generated during the in vitro LDL glycation in incubation mixtures were determined using the cholesterol oxidase (CHOD)-iodide colorimetric assay. 31 The trinitrobenzene sulphonic acid (TNBS) assay was used to measure the amount of free amino group consumption that occurred during in vitro glycation of LDL and HDL. 32 Electrophoretic mobility of the in vitro glycated LDL relative to native LDL was determined using 15µg of native LDL (freshly prepared) or LDL glycated experimentally in the presence or absence of HDL as described previously. 33 Gels were visualized under ultraviolet (UV) light and photographed using a Unipro Platinum gel documentation and analysis system (Uvitec, Cambridge, UK).
Fasting blood glucose was measured in fluoride plasma using the Roche glucose oxidase phenol 4-aminoantipyrine peroxidase (GOD-PAP) method and a Roche auto-analyzer (Roche-Diagnostics Ltd, Burgess Hill, UK) in the Clinical Biochemistry Laboratory of the Manchester Royal Infirmary, which participates in a national quality control scheme. Plasma cholesterol, HDL cholesterol, triglyceride, apoB and apoAI were measured using a Cobas Mira auto-analyzer with HORIBA ABX and Roche standards, control and reagents (HORIBA ABX-UK, Northampton, UK and Roche-Diagnostics Ltd). The assays were calibrated before each test run, as recommended by the manufacturer. PON1 activity was measured in serum samples using paraoxon as substrate. 34
Statistical analyses
Statistical analyses of data were done by the Statistical Package for Social Sciences software (SPSS, Chicago, IL, USA). Results were expressed as mean ± standard error of the mean (SEM) except for triglyceride and PON1 activity, which had a non-Gaussian distribution, for which median and range were used. Statistical differences were sought using Student’s t-test (Wilcoxon rank-sum test was used for triglyceride and PON1 activity) or one-way analysis of variance (ANOVA) followed by Fisher’s least significant difference (LSD) post hoc test (if more than two sets of data were being compared), taking p < 0.05 as statistically significant.
Results
Effect of HDL on glycation of LDL
Serum from 32 participants (20 Lipid Clinic patients and 12 healthy volunteers) was used to investigate the effect of HDL isolated from human serum on autologous LDL glycation. Their clinical characteristics are shown in Table 1. Serum HDL cholesterol ranged from 0.76 to 2.01 mmol/L. The table also shows the data for the same participants divided into those whose serum PON1 activity was above and below the median value for the whole group.
Characteristics of the participants.
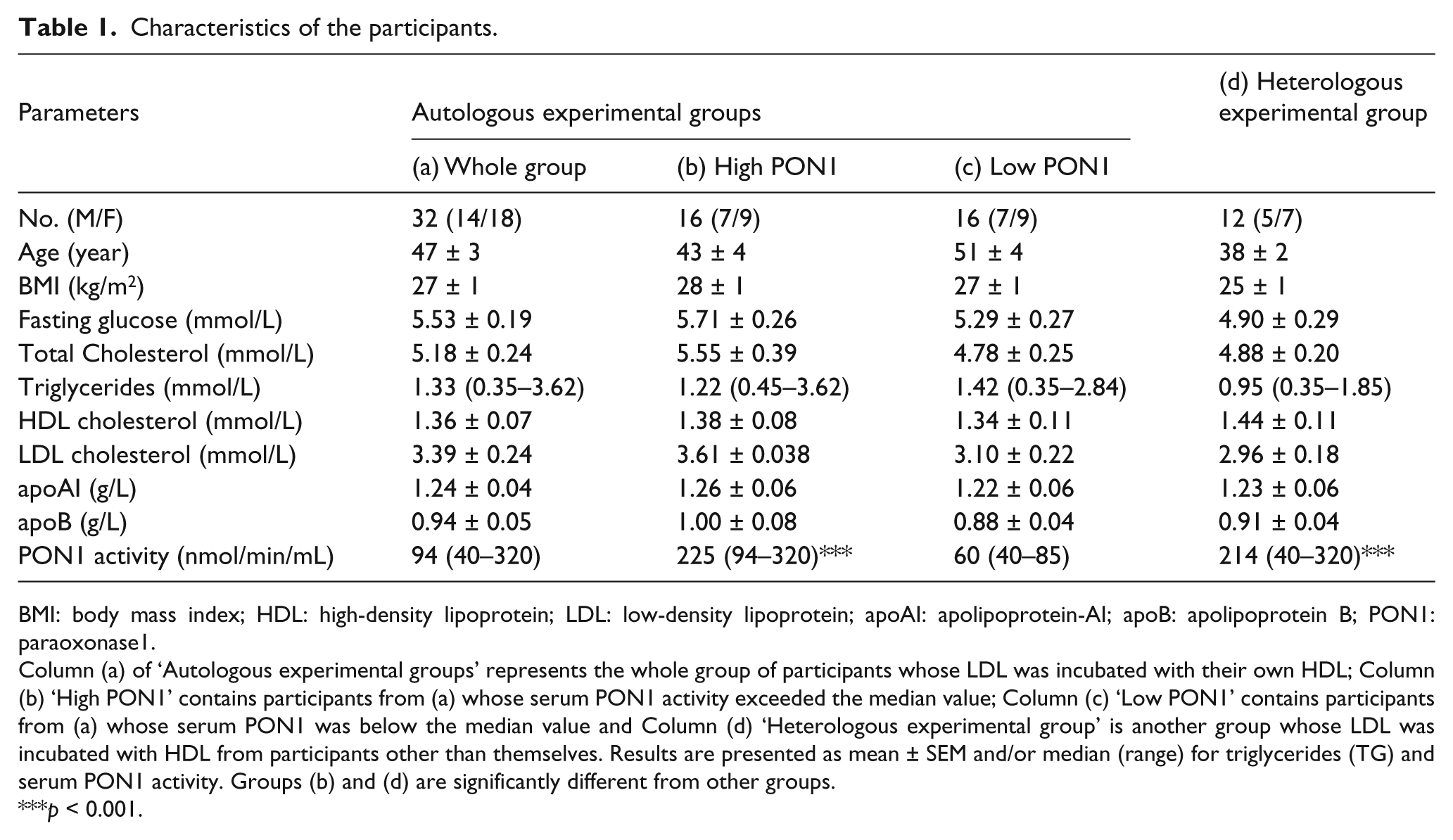
BMI: body mass index; HDL: high-density lipoprotein; LDL: low-density lipoprotein; apoAI: apolipoprotein-AI; apoB: apolipoprotein B; PON1: paraoxonase1.
Column (a) of ‘Autologous experimental groups’ represents the whole group of participants whose LDL was incubated with their own HDL; Column (b) ‘High PON1’ contains participants from (a) whose serum PON1 activity exceeded the median value; Column (c) ‘Low PON1’ contains participants from (a) whose serum PON1 was below the median value and Column (d) ‘Heterologous experimental group’ is another group whose LDL was incubated with HDL from participants other than themselves. Results are presented as mean ± SEM and/or median (range) for triglycerides (TG) and serum PON1 activity. Groups (b) and (d) are significantly different from other groups.
p < 0.001.
On incubation of LDL in the absence of glucose and with glucose 30, 50 and 80 mmol/L for 7 days, its glycated apoB component approximately doubled in the presence of 50 and 80 mmol/L glucose (p < 0.001) (Figure 1). This increase was significantly diminished when autologous HDL was also present (p < 0.05 and p < 0.001, respectively). Glycation of apoAI in HDL, which at baseline was 0.49 ± 0.10% of total apoAI (mean ± SEM), was not significantly increased on incubation with glucose at any of the glucose concentrations used under the conditions of our experiments.
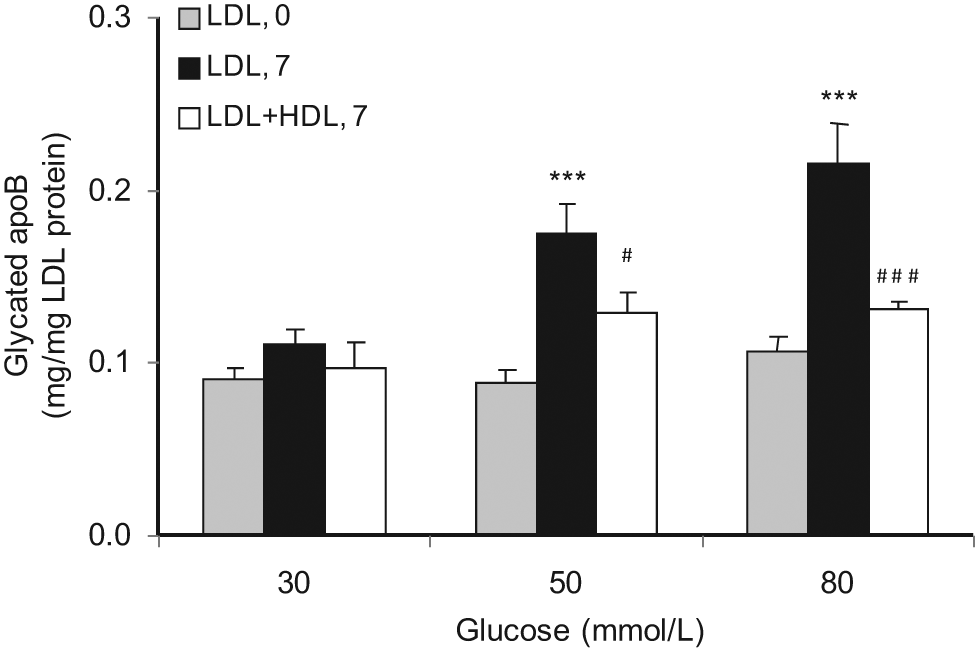
The glycated apoB100 component of human LDL (mg/mg LDL protein) at baseline (LDL, 0) and after in vitro glycation with 30, 50 and 80 mmol/L glucose at 37°C for 7 days either alone (LDL, 7) or in the presence of autologous HDL (LDL + HDL, 7) (n = 32 experiments). Data are presented as mean ± SEM.
Effect of HDL from participants with high and low PON1 activity on LDL glycation
The participants whose serum PON1 activity was below the median (94 nmol/min/mL) (n = 16) were compared with those with values above the median (n = 16) (Figure 2(a) and (b)). Both groups were similar in age and body mass index (BMI) with no significant difference in their fasting blood glucose or lipid profile (Table 1).
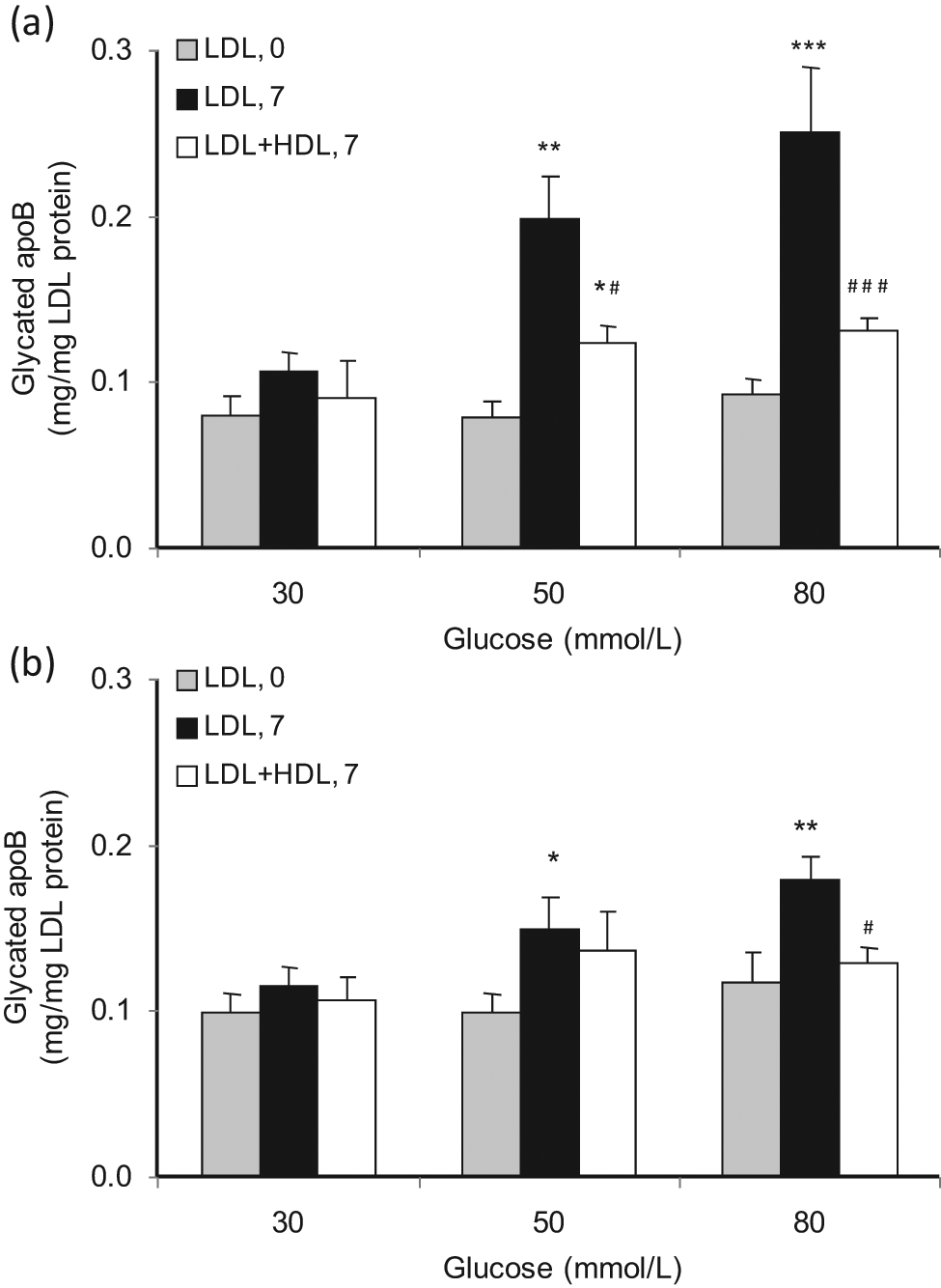
The glycated apoB100 component of human LDL (mg/mg LDL protein) at baseline (LDL, 0) and after in vitro glycation with 30, 50 and 80 mmol/L glucose at 37°C for 7 days either alone (LDL, 7) or in the presence of autologous HDL (LDL + HDL, 7) isolated from volunteers with (a) high serum PON1 activity (n = 16 experiments) and (b) low serum PON1 activity (n = 16 experiments). Data are represented as mean ± SEM.
LDL apoB was protected against glycation by HDL from volunteers with high serum PON1 activity when incubated with glucose 50 and 80 mmol/L (p < 0.05 and p < 0.001, respectively; Figure 2(a)), but HDL from those with low PON1 activity only protected at glucose 80 mmol/L and then to a lesser degree (p < 0.05; Figure 2(b)). LDL apoB glycation was decreased by 25 ± 15% at glucose 50 mmol/L by HDL from those with high PON1 as opposed to 11 ± 13% in those with low PON1, and at glucose 80 mmol/L by 34 ± 7% from those with high PON1 HDL as compared to 22 ± 8% in those with low PON1 HDL (both p < 0.05). Thus, the protective effect of HDL was more pronounced in people with high serum PON1 activity.
In the absence of HDL, there was a tendency for LDL from people with high serum PON1 to be more susceptible to glycation at glucose 50 and 80 mmol/L (Figure 2(a) and (b)). After incubation with glucose 50 mmol/L, glycation of LDL apoB from the low-PON1-activity group was 0.15 ± 0.02 mg/mg protein compared to 0.20 ± 0.03 mg/mg for LDL from the high-PON1-activity group (NS), and with glucose 80 mmol/L, LDL apoB glycation was 0.18 ± 0.02 and 0.25 ± 0.04 mg/mg, respectively (p < 0.01), for the low-PON1-activity and high-PON1-activity groups.
Effect of heterologous HDL on LDL glycation
Cross-over incubation experiments, in which the LDL of 12 healthy volunteers was incubated with 80 mmol/L glucose in the presence of and absence of their own HDL and of HDL from another participant, revealed that heterologous human HDL protected LDL against glycation similar to autologous HDL (Figure 3). Characteristics of the participants whose LDL and HDL were used for cross-over experiments are shown in Table 1. Healthy volunteers, whose serum PON1 activity was typically higher than that of the larger group with its broader spectrum of PON1 activity, participated in these experiments, because, as we had previously shown, HDL from low PON1 serum had little capacity to impede LDL glycation.
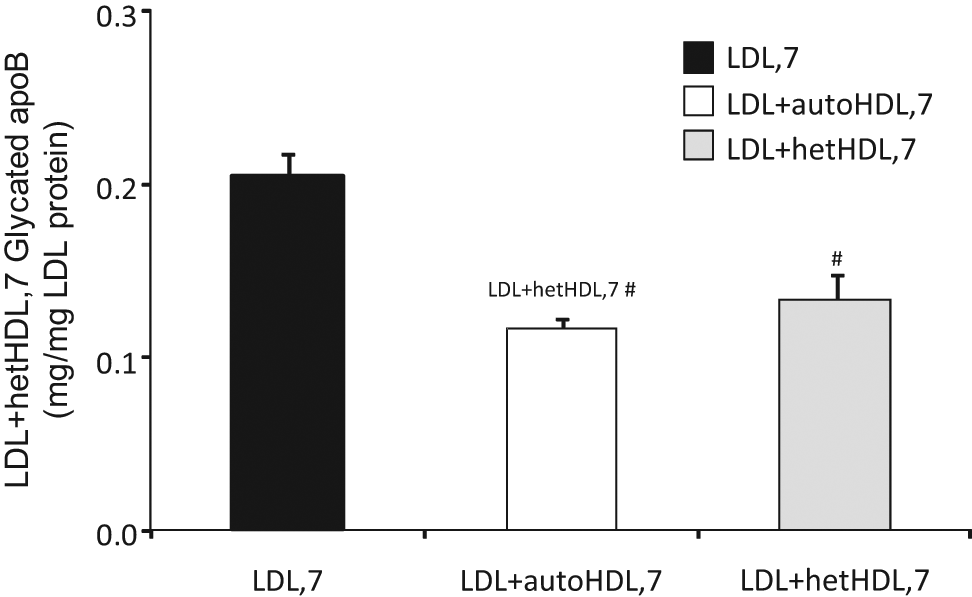
Glycated apoB100 of LDL (mg/mg LDL protein) after in vitro glycation of LDL (n = 12) with 80 mmol/L glucose at 37°C for 7 days either alone (LDL, 7) or in the presence of autologous HDL isolated from the same individual (LDL + autoHDL, 7) or heterologous HDL isolated from another individual (LDL + hetHDL, 7). Data are represented as mean ± SEM.
Effect of HDL on glycation-induced changes in lipid peroxidation, free amino groups and REM of LDL
LPO increased during LDL glycation with 30–80 mmol/L glucose (Figure 4). This increase was largely abolished when autologous HDL was present (Figure 4(a)). This amelioration of LPO accumulation on LDL was mostly evident with HDL from participants with higher serum PON1 activity (Figure 4(b)), but not HDL from those with low serum PON1 activity (Figure 4(c)). HDL from high-PON1- activity participants significantly decreased (p < 0.01) the accumulation of LPO on incubation with heterologous LDL, while that from low-PON1-activity volunteers was as ineffective as it was with autologous LDL.
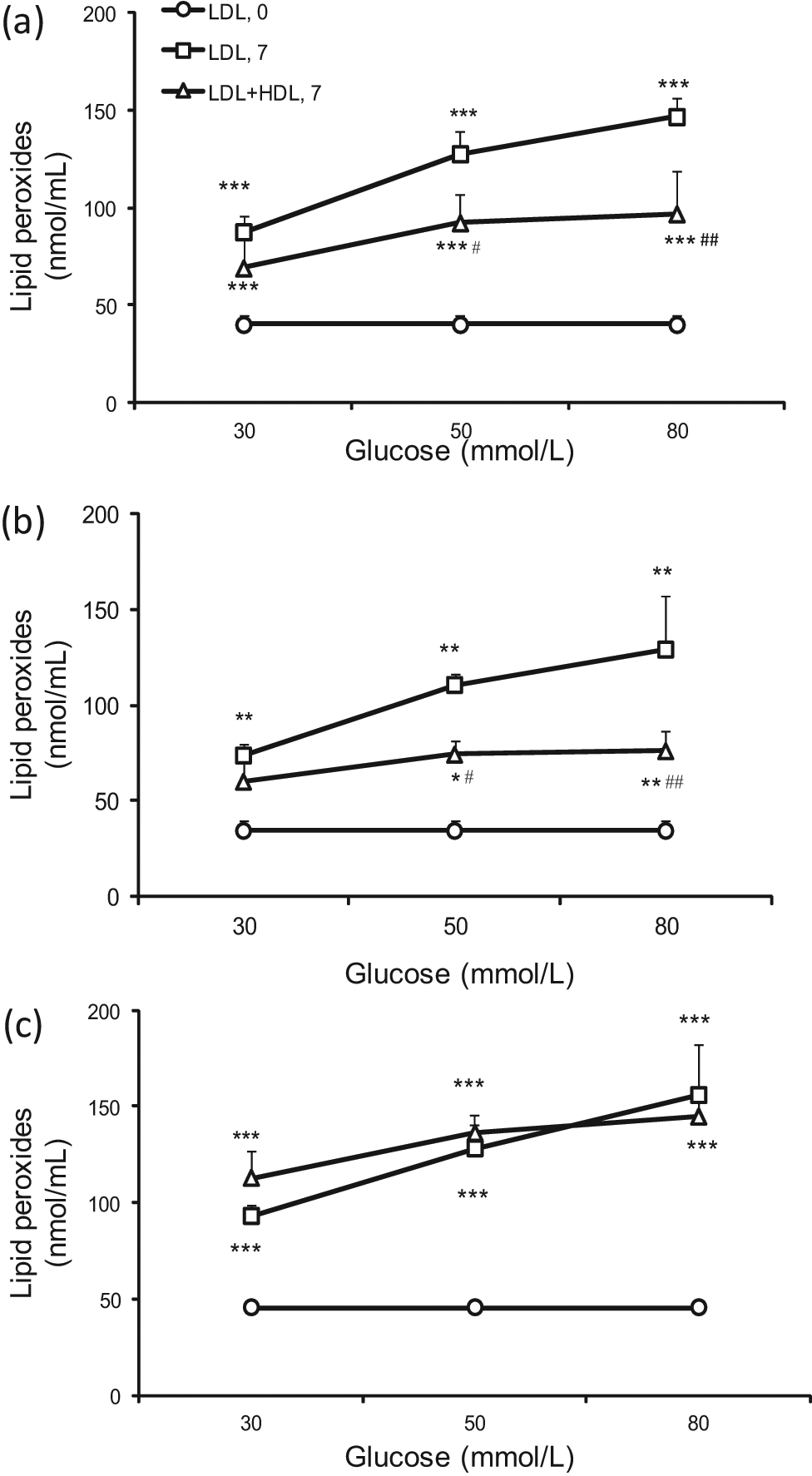
Concentration of lipid peroxides (nmol/mL) in LDL at baseline (LDL, 0) and after in vitro glycation with 30, 50 and 80 mmol/L glucose at 37°C for 7 days either alone (LDL, 7) or in the presence of (a) autologous HDL (LDL + HDL, 7) (n = 20 experiments), (b) the results for HDL from the upper half of the serum PON1 activity frequency distribution (n = 10 experiments) and (c) results from the lower half (n = 10 experiments). Data are represented as mean ± SEM.
The percentage of free amino groups present in LDL protein decreased progressively after incubation of LDL alone with increasing concentrations of glucose: −27 ± 5% at 30 mmol/L, −35 ± 4% at 50 mmol/L and −60 ± 3% at 80 mmol/L. Free amino group loss was significantly lower with LDL in the presence of HDL at glucose 30–80 mmol/L: −11 ± 4% at 30 mmol/L (p < 0.05), −18 ± 5% at 50 mmol/L glucose (p < 0.05) and −24 ± 4% at 80 mmol/L glucose (p < 0.001), respectively. The concentration of free amino groups in LDL from participants with high serum PON1 activity was higher than that in LDL from those with low PON1 activity (0.25 ± 0.03 mg/mL compared to 0.17 ± 0.02 mg/mL), a difference which just failed to achieve statistical significance (p = 0.056). HDL from high-PON1-activity participants decreased the loss of free amino groups of LDL on incubation with glucose significantly (p < 0.05 at glucose 30 and 50 mmol/L and p < 0.001 at glucose 80 mmol/L), whereas HDL from low-PON1-activity volunteers decreased the loss of free amino groups of LDL only with glucose 80 mmol/L (p < 0.01), but not with other glucose concentrations.
LDL isolated from all groups studied showed similar increases in electrophoretic mobility (REM), relative to baseline LDL, after being glycated in vitro for 7 days at 37°C with glucose. Human HDL significantly inhibited the increase in REM of autologous LDL. Although both HDL from participants with low and high serum PON1 activity significantly inhibited the increase in REM of autologous LDL (p < 0.001), HDL with high PON1 activity showed more pronounced inhibition than HDL with low PON1 activity (p < 0.001) (Figure 5). Both autologous and heterologous HDL decreased the REM of LDL after glycation (p < 0.001 for both low- and high-PON1-activity groups). This protective effect was greater for HDL of high PON1 activity (REM at glucose 80 mmol/L, 1.6 ± 0.08, 1.19 ± 0.05 and 1.19 ± 0.05 for LDL without HDL, for LDL with autologous HDL and LDL with heterologous HDL, respectively) than that of low PON1 activity (1.73 ± 0.05, 1.40 ± 0.06 and 1.23 ± 0.05 for LDL without HDL, LDL with autologous HDL and LDL with heterologous HDL, respectively).
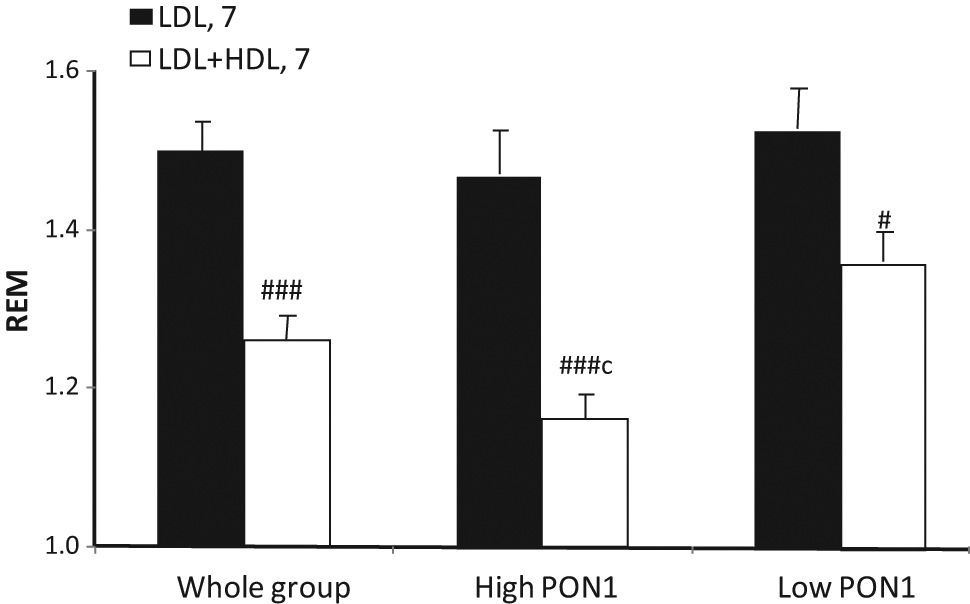
Electrophoretic mobility of LDL relative to baseline (REM) after in vitro glycation with glucose 80 mmol/L in the presence of and absence of autologous HDL from 32 participants (whole group), and the whole group divided into those whose serum PON1 activity exceeded the median and those in whom it was lower (n = 16 in each). Data are represented as mean ± SEM.
Discussion
We report that HDL impedes the glycation of LDL. HDL has been repeatedly reported to decrease LPO accumulation on LDL.3–8,25 Our study is, however, the first report that HDL can directly interfere with the glycation of apoB or indeed any protein. It has been previously reported that in diabetes low circulating levels of HDL components are associated not only with atherosclerosis but also with microvascular complications,12–22 although it is not possible in such studies to determine whether this is due to HDL or some other associated feature of diabetic dyslipidaemia. If HDL has a similar effect in vivo to that reported here, our experiments suggest that HDL might indeed be protective against glycation.
Elucidation of the mechanism by which HDL interferes with LDL glycation remains to be established. However, our results give some indication of potential factors involved. First, HDL itself does not appear to offer itself for glycation in preference to LDL. Indeed, our results reveal that it is more resistant to glycation in vitro than LDL. Second, we provide evidence that PON1 is associated with the anti-glycative capacity of HDL. A previous study has drawn attention to the greater likelihood of microvascular complications in diabetic patients whose HDL is deficient in antioxidative capacity, 18 and we and others have previously found an association between low activity of the PON1 component of HDL or of PON1 genotypes associated with low PON1 activity and diabetic microvascular complications, particularly retinopathy, and with advanced glycation end products.12–14,16,17,19,20 It was for this reason that we examined the effect of HDL varying in PON1 activity. Serum PON1 is almost exclusively located on HDL. 5 Our experiments showed that HDL from people with higher PON1 activity impeded glycation more effectively than that from people with lower activity. Third, by crossing over HDL from healthy people with heterologous LDL we established that protection of LDL against glycation was a general property of HDL when PON1 activity was in the healthy range. There would have been no point in conducting these experiments with HDL from low-PON1-activity individuals, because we had demonstrated that their HDL protected poorly against LDL glycation. Fourth, we noted that the LDL prepared from high-PON1-activity volunteers tended to undergo glycation more extensively. Probably, this was because it possessed more lysine amino groups vulnerable to glycation indicated by its higher free amino group concentration. This was likely to be due to the greater protection afforded to it against glycation by high-PON1-activity HDL before its isolation from the circulation. Physical or compositional variation in LDL from different individuals may influence its capacity to receive protection against glycation by HDL. We have, for example, previously demonstrated that buoyant LDL is less readily glycated than smaller, denser subfractions.33,35
Oxidation is known to accompany glycation, and it has been argued that glycation should more properly be described as glycoxidation. 36 In keeping with this, we have previously reported that when LDL is glycated in our experimental system, small quantities of LPO are also generated. 33 Under the conditions, we impose there is no external source of oxygen free radicals. In the present series of experiments, we report for the first time that HDL decreases the glycation-induced accumulation of LPO on LDL. This complements earlier reports that anti-inflammatory HDL protects LDL against oxidative damage induced, for example, with transition metal cations, 5 lipoxygenase or cultured cells. 37 In vivo, of course, where there are other sources of oxygen free radicals besides those generated by glycation, LDL oxidation is likely to proceed faster than in our experiments. Furthermore, glycation may also proceed faster in vivo, both because of the presence of more reactive glucose metabolites such as methyl glyoxal 38 or gluconolactone 39 and because higher levels of oxygen free radicals may lead to higher rates of lipid peroxidation. The products of lipid peroxidation, such as nonaldehyde (nonanal), adduct to the free amino groups of exposed lysine residues of the apoB of LDL, which are also the site at which glucose combines to form a Schiff’s base in the process of glycation. 10 In the present experiments, the decrease in free amino groups of LDL, probably reflected these processes and their consumption, was substantially diminished by HDL. This effect of HDL was greater when it was isolated from people with higher PON1 activity both with autologous and heterologous LDL. The increase in the REM of LDL, characteristic of alterations in its molecular size and charge when it is fragmented by glycation or oxidation, was also ameliorated when HDL was present and was more evident with HDL from people with higher PON1 activity.
Glycation and oxidation are likely to be intimately interrelated and both will modify proteins implicated in atherogenesis or in microvascular disease. Several lines of evidence implicate PON1 located on HDL in maintaining the antioxidative properties of HDL. PON1 transgenic mice demonstrated an improved protection of LDL against oxidation,40,41 but avian HDL, which lacks PON1 protein failed to protect LDL against Cu2+-induced in vitro oxidation, 42 and HDL from PON1 knockout mice lost its capacity to protect against LDL oxidation induced by a co-culture model of artery wall cells. 43 In clinical studies, the serum concentrations of phospholipid peroxidation derivatives have been shown to correlate inversely with PON1 activity. 9 Although, in populations at low risk, PON1 activity has been equivocal as risk factor for CHD,44–46 in pre-existing CHD and in diabetes, it predicted future cardiovascular events.9,26,27 PON1 appears to be a marker for a several HDL functions regarded as anti-atherogenic in addition to its capacity to protect LDL against oxidative modification, 47 including macrophage cholesterol efflux 48 and endothelial NO production. 49 Our present findings that HDL from people with high PON1 activity is more effective in decreasing LPO accumulation, reducing free amino group consumption and ameliorating the increase in REM induced by glucose suggest that PON1 is involved in protecting LDL against oxidative modification and glycation during glycoxidation. However, despite its clear association with these effects, PON1 may not itself be directly responsible for either of them. Highly purified PON1 has, for example, been reported to lack the capacity to protect LDL against lipid peroxidation while retaining high lactonase activity. 50 Recombinant variants of PON1 in which the catalytic site for lactonase activity has been disrupted by directed evolution lose the capacity to enhance the protection that HDL affords LDL against LPO accumulation. 51 By protecting HDL itself against lactone-induced damage, PON1 may, thus, operate to maintain HDL functions, such as its antioxidative or anti-glycative capacity towards LDL, which could involve other HDL components, for example, apoAI 52 or apoM, 53 more directly.
One limitation of our study is that we could not determine PON1 activity after incubation for 7 days with glucose at 37°C because addition of EDTA and BHT to terminate LPO formation would itself have largely abolished residual PON1 activity. Earlier studies by Hedrick et al. 54 showed that incubation of HDL with glucose for 7 days with glucose at 37°C resulted in a 40% reduction in purified PON1 activity and a 30% reduction of PON1 activity in HDL, indicating that considerable PON1 activity is likely to persist in our experiments.
In the present experiments, LDL glycation was accomplished by incubation for 7 days with glucose 30–80 mmol/L under N2. The relative resistance of LDL to glycation in vitro, indicated by the requirement for its exposure to supraphysiological concentrations of glucose for longer than its physiological circulating half-life, has been consistently reported.33,55–57 In a recent review, 10 earlier investigators reported employing glucose in the range 25–500 mmol/L for periods of up to 28 days and in the presence of elemental oxygen. The implication is that glycation occurs more readily in vivo. As has previously been discussed, glucose metabolites produced in vivo may be more reactive38,39,57,58 or LDL may be more susceptible to glycation if it is concurrently reacting with free radicals present at higher concentrations in vivo. 59 Future study is underway to examine these possibilities and the effect of HDL under these conditions. However, for the present report, the increment in LDL glycation achieved experimentally was similar to the increase observed in clinical diabetes,11,35 and our demonstration that HDL can impede this degree of glycation, albeit achieved with particularly high glucose concentrations, may be regarded as a rigorous test of its capacity to protect LDL against glycation.
We conclude that HDL has the capacity to ameliorate LDL glycation. This may be of potential importance in resistance to atherogenesis. Furthermore, if HDL, which is the predominant lipoprotein in the tissue fluid,60,61 can perform a similar function with other proteins, it may have a role in preventing microvascular complications of diabetes. Our findings point to a possible role of PON1 in the mechanism by which HDL interferes with the glycoxidative modification of LDL. Further research should concentrate on whether this involves the lactonase activity of PON1 directly or indirectly in maintaining the capacities of its other components to impede glycoxidation.
Footnotes
Acknowledgements
We acknowledge support from the Manchester Wellcome Trust Clinical Research Facility and Manchester Biomedical Research Centre.
Funding
This research was funded by the British Heart Foundation and the Lipid Diseases Fund (part of the charitable endowment funds held by Central Manchester University Hospitals NHS Foundation Trust).
Conflicts of interest
Authors have no potential conflicts of interest relevant to this article to be reported.