
Abstract
Background:
This study was designed to compare the efficacy and safety of a fenofibrate/pravastatin 160/40 mg fixed-dose combination plus ezetimibe 10 mg triple therapy and simvastatin 20 mg plus ezetimibe 10 mg dual therapy in patients with type 2 diabetes, mixed hyperlipidaemia and cardiovascular disease.
Method:
After a 6-week run-in period on simvastatin 20 mg, 273 patients with non-high-density lipoprotein cholesterol (non-HDL-C) ≥ 100 mg/dl or low-density lipoprotein cholesterol (LDL-C) ≥ 70 mg/dl were randomised to receive 12-week treatment with triple therapy or dual therapy, followed by a 12-week safety period during which all patients received the triple therapy.
Results:
At week 12, similar significant decreases in non-HDL-C were observed with both treatments. The triple therapy has induced a greater decrease in triglycerides (between-treatment difference: −14.6%, p = 0.007) and the dual therapy a greater decrease in LDL-C (between-treatment difference: +5.3%, p = 0.05). Both treatments were generally well tolerated.
Conclusion:
The fenofibrate/pravastatin plus ezetimibe therapy improves the global atherogenic lipid profile in type 2 diabetic patients with mixed hyperlipidaemia.
Keywords
Introduction
Cardiovascular disease (CVD) is the major cause of both morbidity and mortality in people with type 2 diabetes. Current guidelines recommend reducing levels of low-density lipoprotein cholesterol (LDL-C) as the primary therapeutic target and levels of non-high-density lipoprotein cholesterol (non-HDL-C) as a secondary target for the treatment of diabetic dyslipidaemia.1–4
Statin therapy is recommended for patients with type 2 diabetes and CVD, but their therapeutic needs are likely to exceed LDL-C lowering by statin monotherapy. Indeed a combination therapy should be frequently considered to reach the LDL-C (< 70 mg/dl) and non-HDL-C (< 100 mg/dl) goals. Moreover, elevated triglycerides (TG) and low HDL-C concentrations play a role in the residual cardiovascular risk on statin treatment.5,6
Several options of combination therapy are available for correction of atherogenic diabetic dyslipidaemia. Ezetimibe, when combined with a statin, significantly lowers LDL-C, modestly decreases TG and raises HDL-C levels.7,8 Fenofibrate in combination with a statin induces beneficial effects on the atherogenic lipid profile observed in patients with type 2 diabetes and mixed hyperlipidaemia.9–11 Moreover, the results from the Action to Control Cardiovascular Risk in Diabetes (ACCORD) trial support the use of fenofibrate/statin combination therapy in patients with type 2 diabetes and atherogenic dyslipidaemia defined by high TG and low HDL-C levels. 12 Only few studies have compared the biological effects of fenofibrate (or fenofibric acid) in combination with a statin and ezetimibe (triple therapy), and of statin/ezetimibe dual therapy.13,14
Recently, we have reported that a fixed-dose combination of fenofibrate/pravastatin 160/40 mg improves the atherogenic lipid profile in high-risk patients 15 and in diabetic patients 16 with mixed hyperlipidaemia not controlled by either pravastatin 40 mg 15 or simvastatin 20 mg. 16 But the potential effects of this fenofibrate/pravastatin 160/40 mg fixed-dose therapy used in combination with ezetimibe have not yet been reported in patients with type 2 diabetes not controlled with statin monotherapy.
The objective of this study was to determine whether the efficacy of the fenofibrate/pravastatin 160/40 mg fixed-dose combination therapy plus ezetimibe 10 mg was superior to that of simvastatin 20 mg plus ezetimibe 10 mg combination therapy in decreasing non-HDL-C in type 2 diabetic patients with mixed hyperlipidaemia and cardiovascular disease not at LDL-C and/or non-HDL-C goals on simvastatin 20 mg monotherapy.
Materials and methods
Study design
This multicentre, randomised, double-blind, parallel-group study was conducted according to Good Clinical Practice Guidelines in 73 clinical sites in Europe: in France, 20 sites; Germany, nine sites; Hungary, 10 sites; Norway, eight sites; Poland, 10 sites; and Romania, 16 sites. The study protocol was reviewed and approved by the appropriate ethics committees and institutional review boards in each country, and all patients provided written informed consent before any study procedures were performed. The patients were selected between June 2007 and May 2008 to take part in this study. Laboratoires SMB SA, Brussels, Belgium was the sponsor of the study and covered the cost of all study-related procedures. The contracted research organisation involved in the monitoring was Monitoring Force France, Maisons Laffitte, France.
Eligible patients had to follow the National Cholesterol Education Program (NCEP) Adult Treatment Panel (ATP) III Therapeutic Lifestyle Changes diet 4 for ≥ 12 weeks before the selection visit and be willing to maintain this diet for the duration of the study. Patients were required to discontinue lipid-lowering therapy before entering a 6-week run-in period during which all the selected patients received one tablet of simvastatin 20 mg. Then, eligible patients were randomised in a 1:1 ratio into two groups and took in a double-blind manner either one capsule of fenofibrate/pravastatin 160/40 mg and one tablet of ezetimibe 10 mg or one identical capsule containing one tablet of simvastatin 20 mg and one tablet of ezetimibe 10 mg. Treatments were administered orally during the evening meal for 12 weeks. The 12-week efficacy period was followed by a 12-week open-label safety phase during which all patients received the fenofibrate/pravastatin 160/40 mg capsule and one tablet of ezetimibe 10 mg (Figure 1).
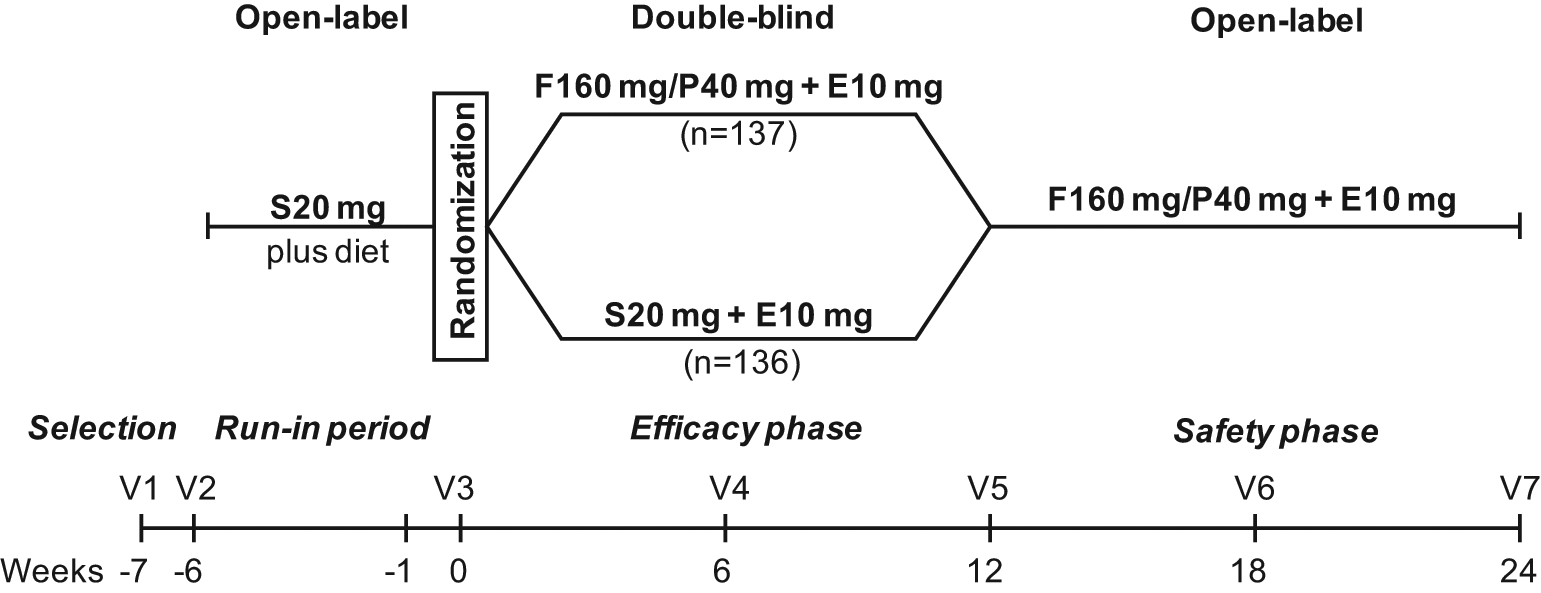
Design of this study of the efficacy and safety of 12-week treatment with fenofibrate/pravastatin 160/40 mg (F160 mg/P40 mg) plus ezetimibe 10 mg (E10 mg) triple therapy versus simvastatin 20 mg (S20 mg) plus E10 mg dual therapy in patients with type 2 diabetes, mixed hyperlipidaemia and cardiovascular disease, uncontrolled with S20 mg. V = visit.
Study population
This study enrolled patients with type 2 diabetes, men and women aged ≥ 18 years, with documented mixed hyperlipidaemia and cardiovascular disease. After the active treatment run-in period with simvastatin 20 mg, type 2 diabetic patients with non-HDL-C ≥ 100 mg/dl or LDL-C ≥ 70 mg/dl and fasting TG ≥ 150 and ≤ 600 mg/dl 1 week before the randomisation visit were randomised (week 0) to either fenofibrate/pravastatin 160/40 mg and ezetimibe 10 mg or simvastatin 20 mg and ezetimibe 10 mg for 12 weeks. The extension safety phase was proposed to all the patients who completed the double-blind period. During the safety phase, all the patients received the fenofibrate/pravastatin 160/40 mg and ezetimibe 10 mg triple therapy for 12 weeks.
Main exclusion criteria were an acute cardiovascular episode within the 3 months before the selection visit; uncontrolled diabetes (glycosylated haemoglobin (HbA1c) > 8.5% 1 week before randomisation) and type 2 diabetes requiring insulin; type I, IIa, IV and V hyperlipidaemia; history of any sensitivity or allergy to statins, fibric acid derivatives and/or ezetimibe; patients with uncontrolled hypertension (systolic blood pressure (BP) > 160 mmHg or diastolic BP > 95 mmHg); history of malignancy; personal or family history of hereditary muscle disease; uncontrolled hypothyroidism; abnormal liver function defined by alanine aminotransferase (ALT) or aspartate aminotransferase (AST) > 2 × the upper limit of normal (ULN) (1.5 × ULN at German sites); creatine kinase (CK) > 3 × ULN (1 × ULN at German sites); creatinine clearance (CrCl) < 60 ml/min and plasma creatinine > 15 mg/l (> 133 μmol/l); use of prohibited concomitant medications; pregnancy or breastfeeding; non-adherence to the NCEP ATP III standardised diet; high alcohol consumption (> 21 beverages per week); and compliance < 80% during the run-in period.
Study procedures
Fasting blood samples were collected at weeks −7, −1, 0 (randomisation), 6, 12 (end of efficacy period), 18 and 24 (end of safety phase) for lipid profile analysis and for clinical chemistry (including ALT, AST, CK and serum creatinine). High-sensitivity C-reactive protein (hs-CRP) and fibrinogen were measured at weeks 0, 6, 12, 18 and 24, homocysteine at weeks 0, 12 and 24, HbA1c at weeks −7, −1, 12 and 24. Serum lipid concentrations were measured using an enzymatic assay (Roche reagents). LDL-C was measured using β-quantification. ApoA-I and ApoB were measured using immunoturbidimetric methodology. All clinical laboratory analyses were performed at a central laboratory (Eurofins Medinet SAS, Plaisir, France).
Safety was assessed by recording the prevalence and severity of adverse events (AEs) and abnormal laboratory data. Each investigator was required to make a causality assessment of the relation of the AE to the study drugs and whether it constituted a serious AE. All study personnel, including investigators, study site personnel, patients, monitors, and central laboratory personnel remained blinded to treatment allocation throughout the efficacy phase of the study. Study personnel remained blinded until the study was completed and the data file was locked.
Outcome measures
The primary efficacy end point was the mean percentage change in non-HDL-C from baseline (i.e. after 6 weeks treatment with simvastatin 20 mg) to the end of the efficacy period. The baseline value was defined as the mean of the measurements obtained 1 week before randomisation and on the day of randomisation. The end-point value was defined as the last post-baseline measurement during the 12-week double-blind efficacy period.
The predefined secondary efficacy end points included percentage changes from baseline to study end point in other lipid and lipoprotein parameters (total cholesterol, LDL-C, HDL-C, TG, apolipoprotein (apo) B, apoA-I, apoB/apoA-I ratio), hs-CRP, fibrinogen and the percentage of patients achieving the ATP III LDL-C goal (i.e. < 70 mg/dl) and/or non-HDL-C goal (i.e. < 100 mg/dl) after 12 weeks of treatment. Other secondary end points were the per cent changes in lipid and lipoprotein parameters at week 24. Safety was assessed by monitoring clinical adverse events (AEs) and vital signs (blood pressure, heart rate and weight at all the visits; ECG at baseline and week 12) in all randomised patients and laboratory AEs (including homocysteine) in all treated patients with ≥ 1 on-treatment measurement available. Pre-specified critical safety parameters included ALT and/or AST elevations > 3 × ULN; CK elevations > 5 × ULN; and creatinine > 20 mg/l (> 177 μmol/l) with ClCr < 50 ml/min. In case of TG concentrations > 600 mg/dl and/or non-HDL-C < 80 mg/dl on two successive assessments, the patient was discontinued according to the investigator’s judgment. Compliance was assessed at each visit by counting the number of returned capsules and tablets.
Statistical analyses
For the sample size calculation, a 5% difference in the non-HDL-C primary efficacy end point was considered as a relevant clinical difference between fenofibrate/pravastatin 160/40 mg plus ezetimibe 10 mg and simvastatin 20 mg plus ezetimibe 10 mg, with a power fixed at 90%. With the hypothesis that standard deviation (SD) of decrease in non-HDL-C would be 11%, 113 usable observations per group were needed (α risk = 0.025, β risk = 0.10, one-sided test). Assuming a potential premature discontinuation rate of 5%, 120 patients were to be included in each group.
Efficacy was evaluated by randomised treatment in the intent-to-treat population, which included all randomly assigned patients with data available for the baseline assessment and at least one post-baseline assessment for the primary end point, and who received at least one dose of active drug during the double-blind period. Analyses used the last-available-observation-carried-forward approach for patients with missing data. A Student t-test for paired series was performed in order to test the changes from baseline to end point in each treatment group. The mean per cent changes from baseline in efficacy parameters were compared between the two treatment groups using an analysis of covariance, which included the baseline value as a co-variable. A p value < 0.025 was considered statistically significant. In cases in which the distribution of the data was non-Gaussian, a non-parametric test was used. The rates of achieving the LDL-C and/or non-HDL-C goals were compared using a logistic regression model. The safety population was defined by all randomised patients who received at least one unit of the study drugs. All the statistics were calculated using SAS version 9.1 (SAS Institute Inc., Cary, North Carolina, USA).
Results
Patient disposition and demographics
The flow of participants through the study is presented in Figure 2. Of 537 patients screened, 273 met the inclusion criteria and were randomised (137 to fenofibrate/pravastatin 160/40 mg plus ezetimibe 10 mg triple therapy and 136 to simvastatin 20 mg plus ezetimibe 10 mg dual therapy). One patient did not receive any dose of study drug, resulting in a safety population of 272 patients for the efficacy period. There were no significant differences in the distribution of baseline demographic, clinical and biological characteristics between the two treatment groups, with the exception of duration of diabetes which was significantly longer in the fenofibrate/pravastatin plus ezetimibe group (p = 0.01) (Table 1). Among the 273 randomised patients, only one patient in the simvastatin plus ezetimibe group had no history of CVD. All the other patients reported at least one CVD. The most frequent history of CVD was angina pectoris (69.5%), followed by myocardial infarction (17.3%), peripheral artery disease (15.1%), revascularisation procedures (13.2%) and stroke (9.2%).
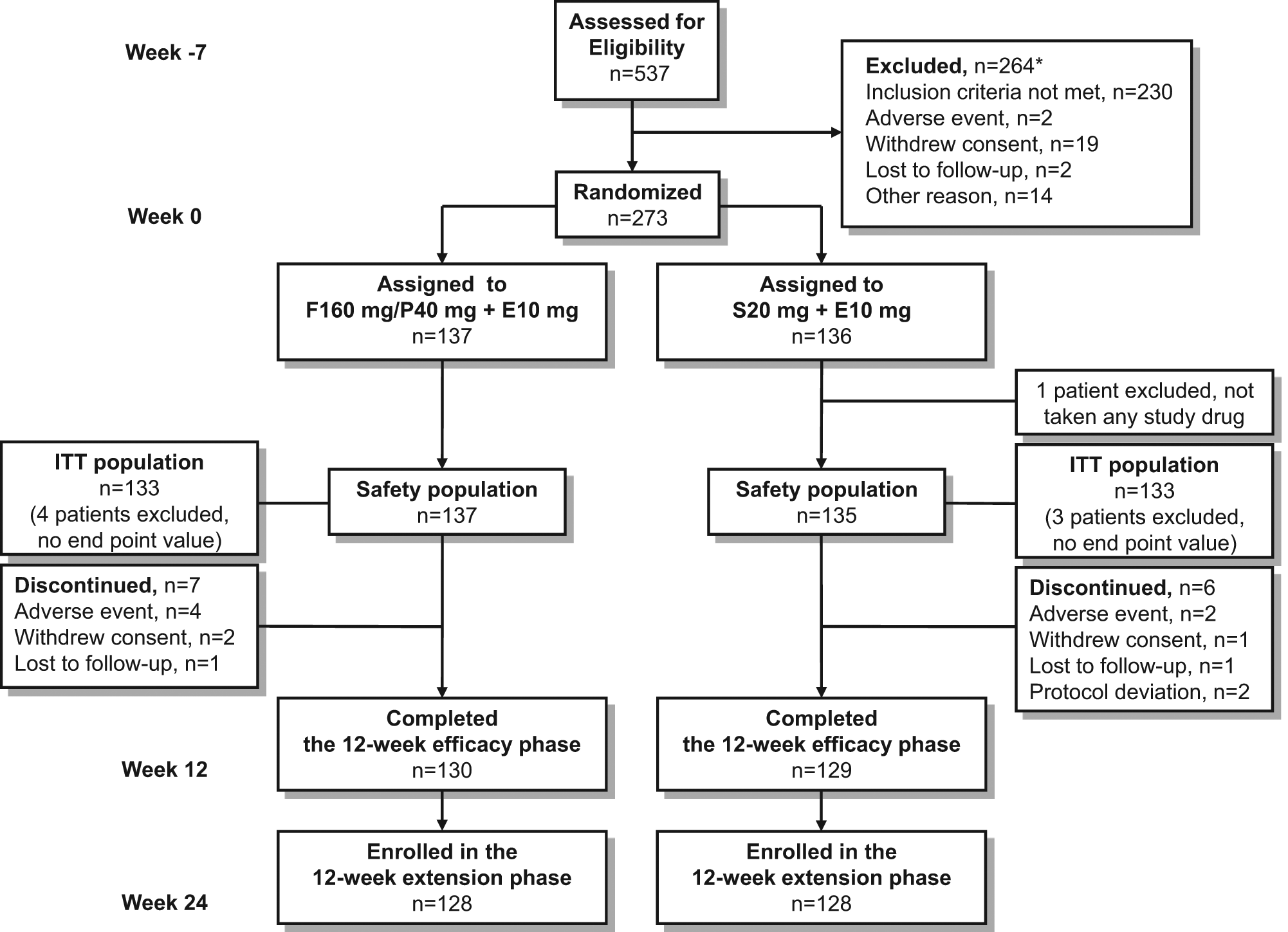
Participant flow chart and statistical analysis sets. E10 mg = ezetimibe 10 mg; F160 mg = fenofibrate 160 mg; ITT = intention-to-treat; P40 mg = pravastatin 40 mg; S20 mg = simvastatin 20 mg. *Some patients reported more than one reason.
Demographics and baseline characteristics (safety population).
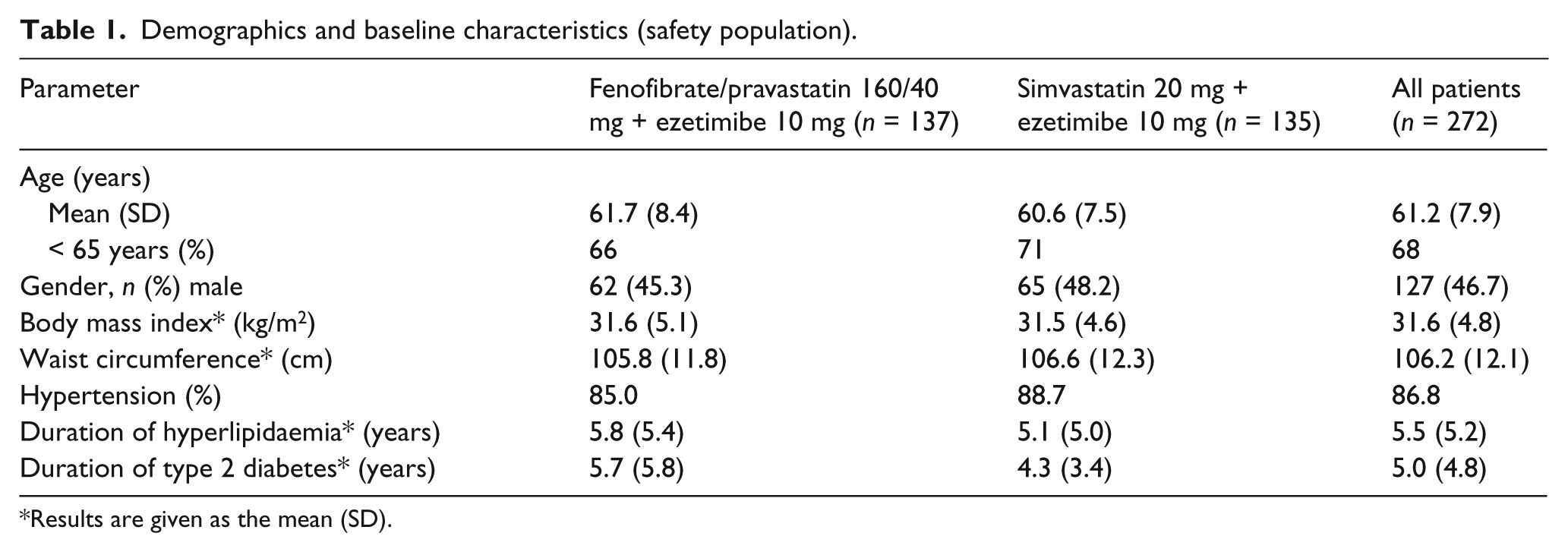
Results are given as the mean (SD).
Among the 266 patients from the intent-to-treat (ITT) population, 51% were receiving statins and 23% fibrates prior to the study and stopped taking it when entering the run-in simvastatin 20 mg phase. The most frequent hypoglycaemic drug was biguanide (75%), followed by sulfonylurea (46%). At the end of the 12-week efficacy study, 100% and 98% of patients treated with, respectively, fenofibrate/pravastatin plus ezetimibe and simvastatin plus ezetimibe had more than 80% compliance by capsule and tablet counts. Among the 259 patients completed the double-blind efficacy phase, 256 entered in the 12-week safety phase. The compliance was also excellent during the safety phase: 99% of patients had more than 80% compliance by capsule and tablet counts. The mean durations of exposure to treatment were 85 and 86 days for, respectively, the efficacy phase and the safety phase.
Efficacy results
The co-administration of either fenofibrate/pravastatin 160/40 mg plus ezetimibe 10 mg or simvastatin 20 mg plus ezetimibe 10 mg resulted in a significant reduction in non-HDL-C after 12 weeks of treatment (p < 0.0001 vs. baseline on simvastatin 20 mg), but with no significant difference between the two groups (between-treatment difference, +3.6%; p = 0.089) (Table 2). In both treatment groups, non-HDL-C levels decreased during the first 6 weeks of treatment and then remained stable until the end of the efficacy phase. At week 12, improvement in TG concentrations was significantly greater with the fenofibrate/pravastatin plus ezetimibe triple therapy compared with simvastatin plus ezetimibe dual therapy (between-treatment difference, −14.6%; p = 0.007). The increase in HDL-C levels was only significant in the fenofibrate/pravastatin plus ezetimibe group (+3.5%; p = 0.03), without significant difference in the HDL-C changes between the two groups (p = 0.066). The same trend was observed in the evolution of apoA-I levels with a small, but significant increase in the fenofibrate/pravastatin plus ezetimibe group (+3.0%; p = 0.01), without significant difference between the two treatment groups (p = 0.063). The simvastatin/ezetimibe combination treatment induced a greater decrease in LDL-C level (−25.1%; p < 0.0001) compared to the decrease observed in the fenofibrate/pravastatin plus ezetimibe group (−19.8%; p < 0.0001), with a significant between-treatment difference of 5.3% (p = 0.05) (table 2). Changes in total cholesterol and apoB concentrations were not significantly different between the two groups. After 12 weeks of treatment, fibrinogen concentration significantly decreased in the fenofibrate/pravastatin plus ezetimibe group, with a significant between-treatment difference (p < 0.0001) (Table 2). The hs-CRP concentrations were not normally distributed and the reported analysis was only descriptive.
Percentage changes from baseline (week 0) in lipid and lipoprotein parameters, fibrinogen, and high-sensitivity C-reactive protein after 12 weeks of treatment (intention-to-treat population).
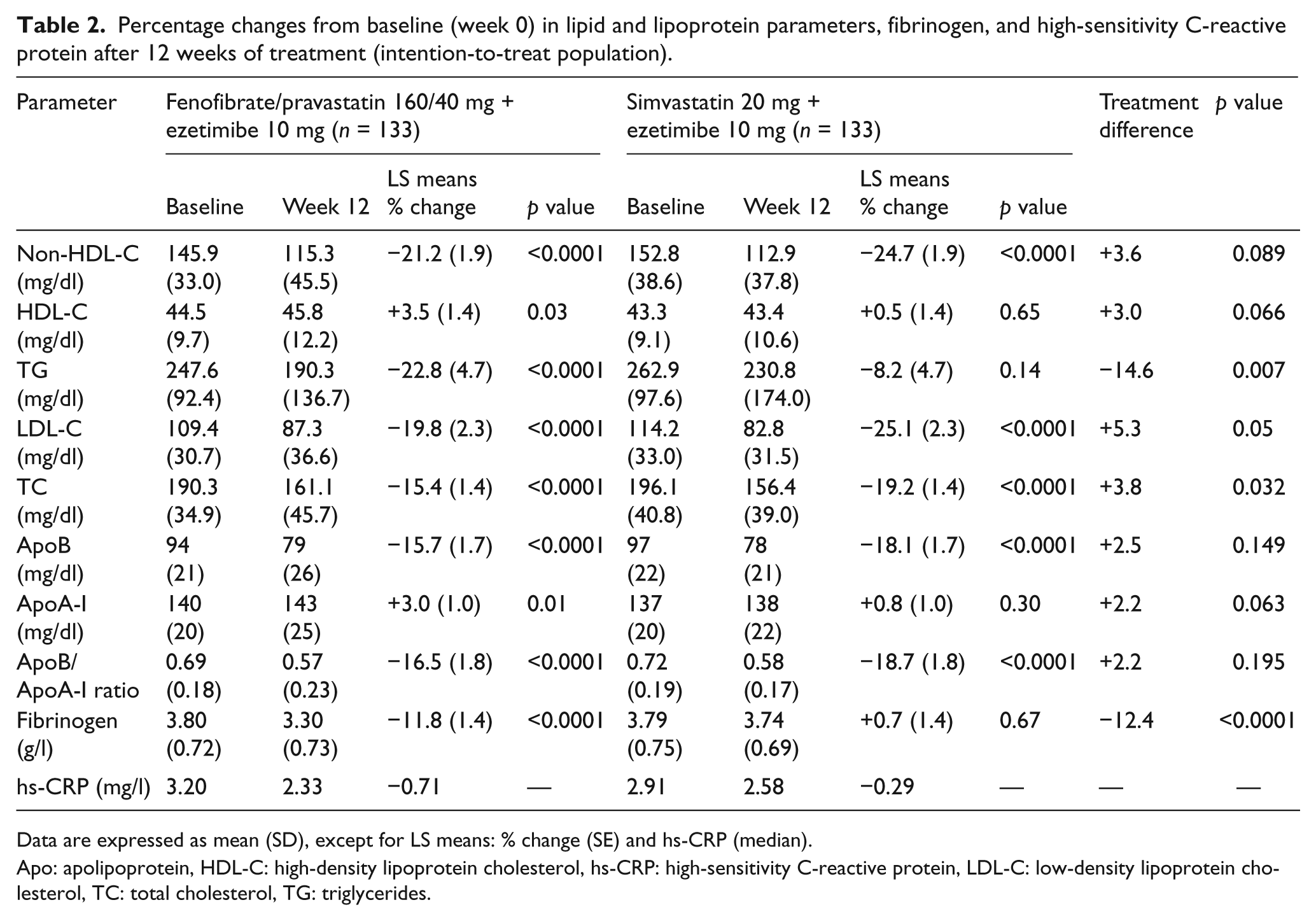
Data are expressed as mean (SD), except for LS means: % change (SE) and hs-CRP (median).
Apo: apolipoprotein, HDL-C: high-density lipoprotein cholesterol, hs-CRP: high-sensitivity C-reactive protein, LDL-C: low-density lipoprotein cholesterol, TC: total cholesterol, TG: triglycerides.
The proportions of patients who achieved the ATP III LDL-C goal (<70 mg/dl) and/or non-HDL-C goal (<100 mg/dl) were not significantly different between the two groups (Figure 3).
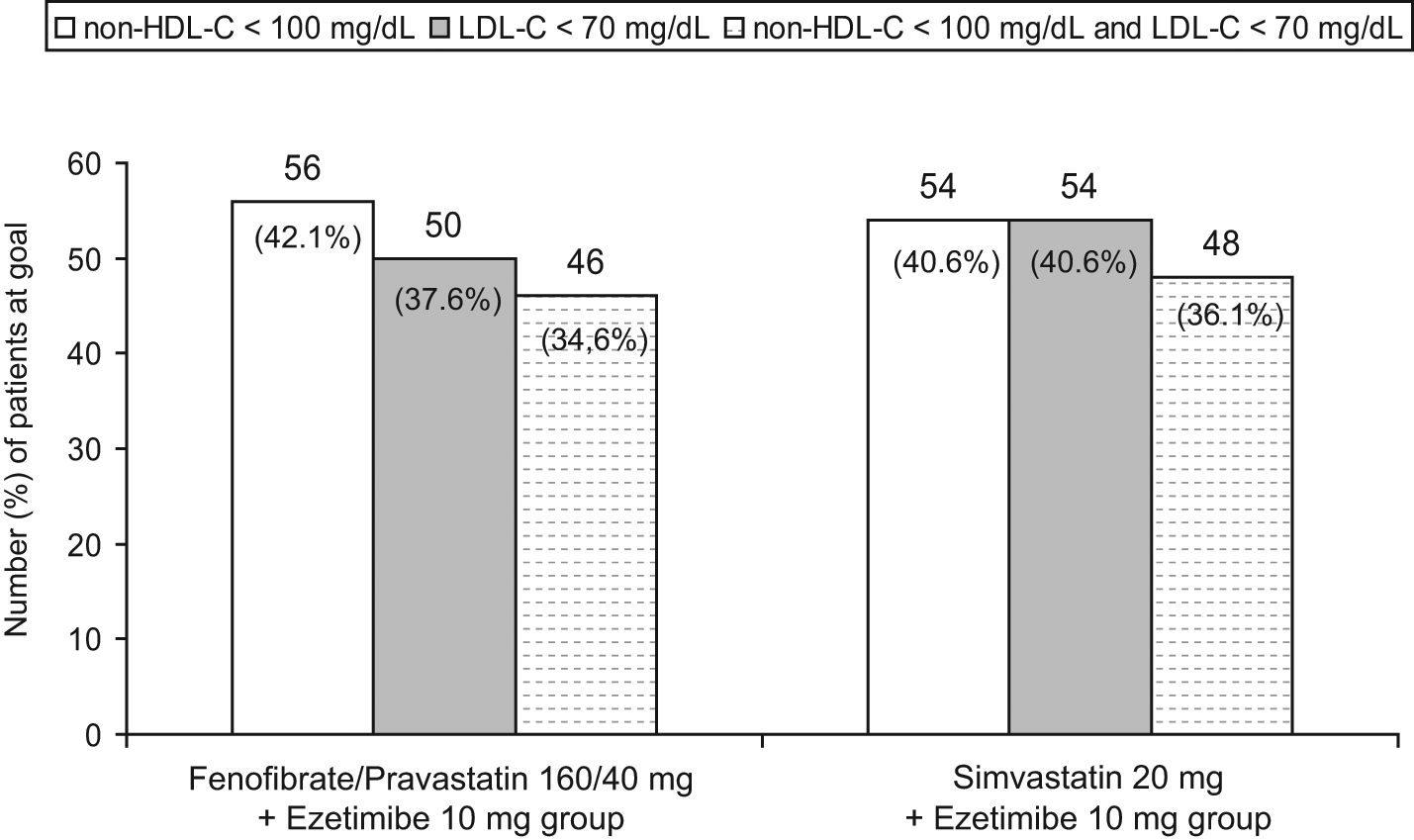
Rates of achievement of the NCEP ATP III therapeutic goals at the end of the efficacy phase (intention-to-treat population).
At the end of the safety extension phase during which all patients received the fenofibrate/pravastatin plus ezetimibe triple therapy, the decrease in non-HDL-C was sustained over 24 weeks and similar both in patients treated with the triple therapy from the beginning of the study and in patients switching from simvastatin plus ezetimibe to triple therapy (Table 3). Globally, the results observed at week 24 are consistent with those obtained during the efficacy phase: compared with the baseline lipid levels observed on simvastatin 20 mg at the end of the run-in period, the fenofibrate/pravastatin plus ezetimibe triple therapy induced significant decreases in non-HDL-C, TG, LDL-C, apoB atherogenic parameters and significant increases in HDL-C and apoA-I levels, associated with a significant decrease in apoB/apoA-I ratio (Table 3).
Percentage changes from baseline (week 0) in lipid and lipoprotein parameters, fibrinogen, and high-sensitivity C-reactive protein at the end of the safety phase (week 24).
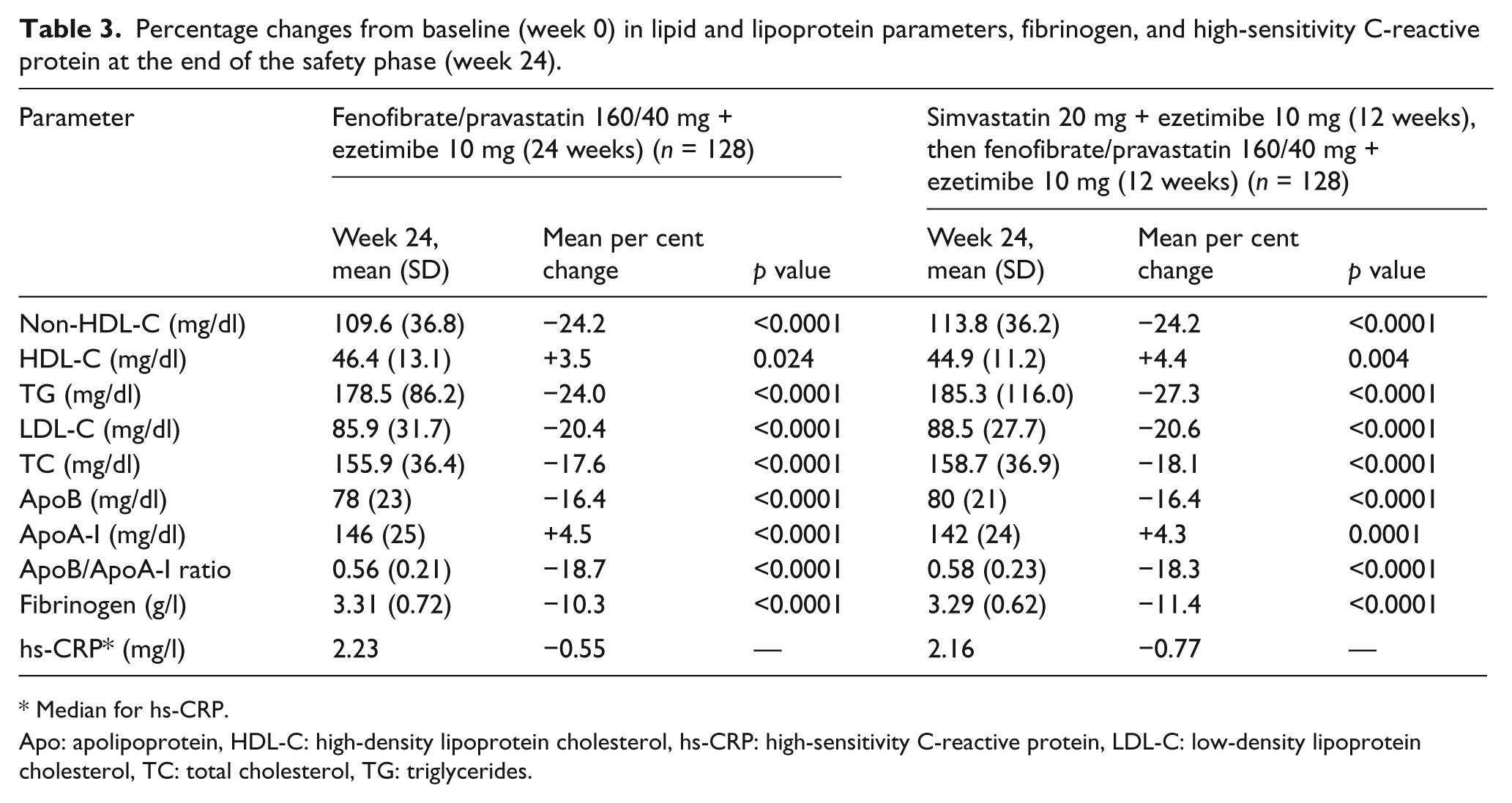
Median for hs-CRP.
Apo: apolipoprotein, HDL-C: high-density lipoprotein cholesterol, hs-CRP: high-sensitivity C-reactive protein, LDL-C: low-density lipoprotein cholesterol, TC: total cholesterol, TG: triglycerides.
Safety results
Clinical adverse events
During the double-blind treatment period, the proportions of patients in whom ≥ 1 AE was reported were not significantly different between the fenofibrate/pravastatin plus ezetimibe and the simvastatin plus ezetimibe groups (14.6% vs. 14.1%) (Table 4). Most of the AEs were considered mild to moderate and no deaths were reported. The frequency of treatment-related AEs was 6.6% in the fenofibrate/pravastatin plus ezetimibe group and 3.0% in the simvastatin plus ezetimibe group, but the difference between groups was not significant (p = 0.16). Serious AEs were reported in four patients (cerebral concussion secondary to a road accident in one patient in the simvastatin plus ezetimibe group; unstable angina in one patient in the fenofibrate/pravastatin plus ezetimibe group; post-operative infection secondary to a planned shoulder surgery in one patient in the fenofibrate/pravastatin plus ezetimibe group; and gastrointestinal haemorrhage and subsequent anaemia in one patient in the fenofibrate/pravastatin plus ezetimibe group, serious AEs considered as treatment-related). Four patients in the fenofibrate/pravastatin plus ezetimibe group and two patients in the simvastatin plus ezetimibe group were discontinued from the study due to AEs related to the study drug.
Number of patients (%) with adverse events (AEs) during the efficacy and safety treatment periods (safety population).
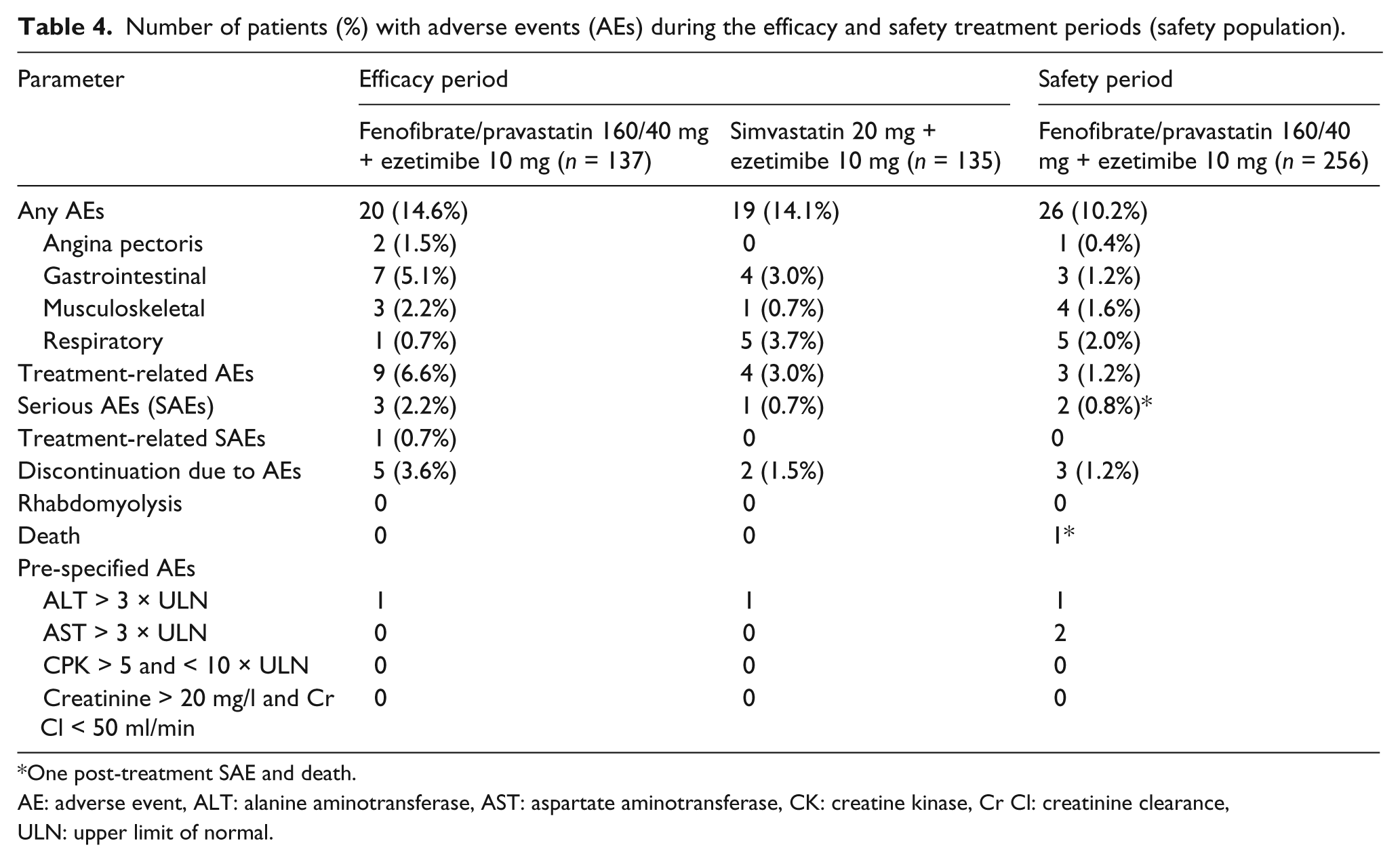
One post-treatment SAE and death.
AE: adverse event, ALT: alanine aminotransferase, AST: aspartate aminotransferase, CK: creatine kinase, Cr Cl: creatinine clearance, ULN: upper limit of normal.
Among the 256 patients enrolled in the safety phase, 26 (10.2%) experienced ≥1 AE from week 12 to week 24 (Table 4). Three AEs were considered treatment related. Most AEs were mild to moderate. Three serious AEs in two patients have been reported during the safety phase: transient ischaemic attack in one patient, serious AEs considered not related to study drug; lymphoma diagnosed more than 30 days after the last study drug intake in one patient for which a second serious AE (acute coronary syndrome and death) occurred also more than 30 days after the last study drug intake, serious AEs considered not related to study drug. There were no cases of myopathy or rhabdomyolysis during the 24-week study period.
Laboratory tests
During the double-blind period, the mean AST level was increased in both groups (+14.5% in the fenofibrate/pravastatin plus ezetimibe group and +9.1% in the simvastatin plus ezetimibe group), without significant between-treatment difference (p = 0.18). The mean ALT level was also increased in the fenofibrate/pravastatin plus ezetimibe group (+5.9%) and in the simvastatin plus ezetimibe group (+11.5%), without significant between-treatment difference (p = 0.12). One patient in each group had an isolated increased of ALT > 3 × ULN, discovered at week 6, resolved at the next visit, and with no concurrent elevation in bilirubin. During the safety phase, an increase in AST or ALT > 3 × ULN was observed in two patients, without a concurrent elevation in bilirubin (Table 4).
The mean CK level was increased in both groups (+14.1% in the fenofibrate/pravastatin plus ezetimibe group and +7.7% in the simvastatin plus ezetimibe group), without significant between-treatment difference (p = 0.18). No patient reported a CK increase > 5 × ULN during either the efficacy or the safety periods.
Mean baseline serum creatinine concentrations and CrCl were respectively 89.3 μmol/l and 90.6 ml/min in the fenofibrate/pravastatin plus ezetimibe group and 87.3 μmol/l and 95.0 ml/min in the simvastatin plus ezetimibe group. At week 12, creatinine concentrations were significantly increased in both the fenofibrate/pravastatin plus ezetimibe group and the simvastatin plus ezetimibe group (by 13.5% and 4.4%, respectively; p < 0.0001 between groups). Significant decreases in CrCl were observed in both the fenofibrate/pravastatin plus ezetimibe (−10.3%) and the simvastatin plus ezetimibe group (−3.0%), with a significant (p < 0.0001) between treatment difference. No patients had the pre-specified AE creatinine concentration > 20 mg/l and CrCl < 50 ml/min. The homocysteine concentrations were not significantly changed from baseline in the simvastatin plus ezetimibe group (+2.8%, p = 0.27), but were significantly increased (+28.5%, p < 0.0001) in the fenofibrate/pravastatin plus ezetimibe group (p < 0.0001 between groups). Any relevant changes between groups were observed neither in fasting blood glucose nor in HbA1c.
Discussion
Patients with type 2 diabetes and CVD are at very high risk of recurrent cardiovascular events. Diabetic dyslipidaemia is strongly related to this increased CVD risk and lipid-lowering treatment appears to account for the major reduction of CVD events. 17 Statins have proved to be highly effective in reducing the levels of LDL-C leading to significant reductions in the incidence of CVD events. 18 However, despite statin therapy, high CVD risk persists in these patients and a more-global approach to lipid management is likely to result in further risk reduction. Diabetic dyslipidaemia is indeed characterised by an atherogenic lipid profile including elevation of TG concentration, low HDL-C and apoA-I levels, excess of small dense LDL and increases of non-HDL-C and apoB levels. Combinations of lipid-lowering drugs are frequently required to achieve guideline-specified goals in patients with type 2 diabetes and CVD.1–4
The objective of this randomised, double-blind study was to compare the efficacy and safety profile of fenofibrate/pravastatin 160/40 mg plus ezetimibe 10 mg (triple therapy) and simvastatin 20 mg plus ezetimibe 10 mg (dual therapy) in patients with type 2 diabetes and mixed hyperlipidaemia and with cardiovascular disease in whom LDL-C goal or non-HDL-C goal has not been achieved with simvastatin 20 mg monotherapy. The use of a fenofibrate/pravastatin 160/40 mg fixed dose combination was supported by the limited pharmacokinetic interaction between fenofibrate and pravastatin19,20 and by the efficacy and safety profile observed with this fixed dose combination in two previous trials.15,16 Ezetimibe has a complementary mechanism of action to the statins and thus ezetimibe in combination with a statin has been shown to induce a 15–20% complementary reduction in LDL-C and non-HDL-C (or apoB). A recent pooled analysis has also suggested that ezetimibe/statin combination therapy could induce larger reductions in LDL-C and non-HDL-C in patients with diabetes than those without diabetes. 8 Moreover, ezetimibe did not significantly affect fenofibrate pharmacokinetics, and fenofibrate increased steady state exposure of ezetimibe which was not considered clinically relevant. 21
In this study, the use of fenofibrate/pravastatin 160/40 mg plus ezetimibe 10 mg triple therapy was not associated with a significantly greater reduction in non-HDL-C (primary end point) compared with simvastatin 20 mg plus ezetimibe 10 mg dual therapy. But both strategies have induced a highly significant complementary decrease in non-HDL-C and in the other atherogenic parameters (LDL-C, total cholesterol and apoB). The dual therapy was more effective in reducing LDL-C. In contrast, the triple therapy was associated with statistically significant beneficial effects on HDL-C, apoA-I and TG concentrations, the between-treatment difference reaching significance only for TG levels. These findings are consistent with data from a previously published study that compared ezetimibe/simvastatin plus fenofibrate and ezetimibe/simvastatin treatments in patients with mixed hyperlipidaemia. 13 By comparison to dual therapy, the fenofibrate/pravastatin plus ezetimibe triple therapy mainly differ in the potential to modulate the atherogenic lipid profile by concomitantly lowering TG and raising HDL-C levels. However, this trial confirmed that the HDL-C increasing effect is markedly less in patients with type 2 diabetes, as observed in long-term intervention trials.12,22 Globally, with the significant decreases in LDL-C and non-HDL-C levels observed with both treatment strategies, the proportion of patients achieving the LDL-C and/or non-HDL-C goals were similar with either fenofibrate/pravastatin plus ezetimibe or simvastatin plus ezetimibe therapies. The overall efficacy of the triple therapy was confirmed at the end of the safety phase.
Safety concerns, especially with respect to muscle-related safety, have initially restricted the clinical use of combination therapy with a fibrate and a statin.23,24 A recent retrospective pharmacoepidemiology safety study has confirmed an increased risk of myopathy and rhabdomyolysis with fibrate plus statin combination therapy. 25 However there are differences in the prevalence of muscular risk between fibrates: in combination with a statin, fenofibrate has been reported to be associated with fewer pharmacodynamic and pharmacokinetic interactions compared with gemfibrozil, 26 and to be consistently better tolerated than gemfibrozil.23,24 Moreover fenofibrate administered with simvastatin was well tolerated in the long-term ACCORD study. 12 In this 24-week study, the overall safety and tolerability of the fenofibrate/pravastatin plus ezetimibe triple therapy was good, with no report of myopathy and rhabdomyolysis. The fenofibrate/pravastatin plus ezetimibe triple therapy was associated with an increase in creatinine levels and a decrease in CrCl. These findings were consistent with those observed with fenofibrate treatment.12,22,27 The triple therapy was also associated with a significant increase in homocysteine levels, effect suggested as the basis of the small effect of fenofibrate on HDL-C and apoA-I. 28 The clinical significance of this effect on homocysteine, as well as on fibrinogen, remains unclear.
The major limitations of this study are short duration and insufficient power to assess clinical events. However, findings from the lipid arm in the ACCORD trial have supported the use of a fenofibrate and statin combination therapy in patients with type 2 diabetes and mixed hyperlipidaemia. 12 Moreover in meta-analysis, fibrates had a beneficial effect on major cardiovascular events in high-risk patients with mixed dyslipidaemia. 29 Ezetimibe/simvastatin combination therapy has been also recently shown to reduce the incidence of major atherosclerotic events in patients with chronic kidney disease. 30 Another limitation of this trial is the lack of evaluation of the tested treatments on lipoprotein sub-classes. In a previous study, 31 the ezetimibe/simvastatin plus fenofibrate therapy was found to be effective in moving LDL size pattern from small dense to more larger buoyant LDL particles.
Conclusion
In this population of patients with type 2 diabetes, mixed hyperlipidaemia and cardiovascular disease not controlled by simvastatin 20 mg monotherapy, the fenofibrate/pravastatin 160/40 mg and ezetimibe 10 mg triple therapy and the simvastatin 20 mg plus ezetimibe 10 mg dual therapy were effective at reducing non-HDL-C, LDL-C, TC and apoB levels. The triple therapy induced significant decrease in TG levels and increases in HDL-C and apoA-I levels, with a between-treatment difference only significant for TG concentration. The safety and tolerability profile of triple therapy was similar to that observed with dual therapy. Further studies are needed to determine whether the global improvement in the atherogenic lipid profile obtained with the fenofibrate/pravastatin and ezetimibe therapy seen in this study will lead to decrease the incidence of cardiovascular events in patients with type 2 diabetes and mixed hyperlipidaemia.
Footnotes
Acknowledgements
The authors acknowledge the investigators who participated in this study: from
Funding
This study was financially supported by Laboratoires SMB SA, Brussels, Belgium.
Conflict of interest statement
Dr Farnier has received grant/research support and speaker’s honoraria from and been a consultant and advisor for Abbott, AstraZeneca, Boehringer–Ingelheim, Genzyme, Kowa, Laboratoires SMB, Lilly, Merck & Co, Merck/Schering–Plough, Novartis, Pfizer, Recordati, Roche and Sanofi–Aventis. Dr Steinmetz has received honoraria for lectures or participation in the advisory boards of Astra–Zeneca, Bristol–Myers Squibb Company, Eli Lilly and Company, Merck KGaA, Merck Sharp & Dohme Corp., Novartis, Solvay, and Sanofi–Aventis. The authors have indicated that they have no other conflicts of interest regarding the content of this article.