
Abstract
Because of the current COVID-19-pandemic, the world is currently being held hostage in various lockdowns. ACE2 facilitates SARS-CoV-2 cell-entry, and is at the very center of several pathophysiological pathways regarding the RAAS, CS, KKS, T2DM, and IL-6. Their interactions with severe COVID-19 complications (e.g. ARDS and thrombosis), and potential therapeutic targets for pharmacological intervention, will be reviewed.
Keywords
Introduction
The aim of this review is to create a framework of different interconnected pathways that constitute the complex COVID-19 pathophysiological crossroads, as well as the interactions with common comorbidities (T2DM in particular).
First the SARS-CoV-2 virus and COVID-19 (including severe complications) will be introduced. Then, the COVID-19 relation with the RAAS will be reviewed, and a preliminary framework will be established. This framework will subsequently be expanded with the CS and the KKS. The interactions between COVID-19, T2DM, and IL-6, will be reviewed in light of this framework. Finally, some potential targets for therapeutic intervention will be discussed.
Abbreviations are listed at the end.
COVID-19
SARS-CoV-2 is a SSRNA+, enveloped virus from the beta-coronavirus family, with a structural surface spike (S) glycoprotein that primarily binds to the N-terminal domain of ACE2, predominantly, but not exclusively, on type-II pneumocytes.1–3 SARS-CoV-2 decreases surface ACE2 expression through directly binding to ACE2,4–6 followed by TMPRSS2-mediated proteolytic cleavage3,7,8 and subsequent endocytosis.1,3,5 TMPRSS2 is essential for SARS-CoV-2 membrane fusion and subsequent cell-entry,3,7,8 but other proteases (such as Furin) may facilitate SARS-CoV-2 cell-entry as well.2,5,9 Surface ACE2 is further reduced via shedding, caused by Ang-II-mediated upregulation of ADAM-175,6,10 and androgen-mediated8,11 upregulation of TMPRSS2.3,7 Reduced ACE2 expression may also be modulated epigenetically via DNA-methylation processes,12,13 or via crosstalk with T2DM-induced hyperglycemia.14,15
The SARS-CoV-2-related disease is commonly referred to as COVID-19.1,7,16 Common COVID-19 symptoms are fever, dry cough, sore throat, dyspnea, headache, and myalgia.1,12,17 Some atypical symptoms are anosmia (loss of smell)18–20 and diarrhea (which may present earlier than respiratory conditions).1,5,17 Different sex and age groups have very biased severity and mortality of COVID-19, with male, old age, and comorbidity being the most affected.1,4,15 T2DM, hypertension, and CVD are common COVID-19 comorbidities (which are likely related to a dysregulated RAAS)5,6,21 and are associated with ARDS15,22,23 and high fatality.1,23,24 Currently, there is no approved and effective medication against COVID-19.16,22,25
The protective mechanism during the early stages of a (viral) infection predominantly occurs through the innate immune system.26–28 COVID-19 primarily suppresses the innate immune system, enabling the uncontrolled spread of the virus during the initial stages.27–29 This explains a mild (sometimes even asymptomatic) presentation early on.27,28,30 The high effectiveness of the innate immune system in children possibly explains why COVID-19 doesn’t seem to affect them as much (if at all).27,28,30 Differences in immunity27,28,31 and gene expression4,12,22 may contribute to COVID-19 severity. Severe COVID-19 complications are associated with excessive and dysregulated host immune responses, which may contribute to the development of lethal CRS and ARDS.32–34
Acute respiratory distress syndrome
ARDS, the most severe form of ALI,35–37 is a clinical syndrome of noncardiogenic pulmonary edema.38–40 ARDS is characterized by an excessive inflammatory response,41–43 damage to both alveolar epithelial38,40,41 and vascular endothelial39,44,45 cells, the subsequent breakdown of the alveolar-capillary barrier integrity,38,42,46 impaired AFC,40,41,45 excessive interstitial and parenchymal neutrophil migration,38,43,45 and activation of alveolar macrophages, platelets, and pro-coagulant processes.42,47,48 This may result in diffuse alveolar damage,42,49 pulmonary fibrosis,37,50 and impaired gas exchange,42,51 leading to (refractory) hypoxemia35,38,42 and possibly organ dysfunction.40,47,52
Uncontrolled inflammation leads to excessive and prolonged activation of neutrophils,38,43,45 which are immune cells that play an important role in the regulation of IL-6 signaling53–55 in the pathology of pulmonary inflammatory disorders.38,43,56 Neutrophil-mediated ROS production (via NADPH oxidase),57,58 plays an essential part in the immune response against pathogens via NET formation and direct cellular damage.59–61 Excessive neutrophil recruitment therefore contributes significantly to the severity of inflammatory pneumonia.56,60,62
In AFC, alveolar fluid is cleared via an ENaC-mediated osmotic gradient.40,41,63 Disruption of ENaCs can lead to impaired AFC.42,62,64
The ECM is important for the epithelial and endothelial barrier function, since it regulates intercellular interactions and controls the migration of fluid and molecules in the interstitial space.42,65 Changes in ECM composition affect the mechanical properties of tight-junctions in alveolar epithelial and vascular endothelial cells, modulating the alveolar-capillary barrier function.42,66 Excess deposition of ECM proteins can lead to pulmonary fibrosis,37,42,50 which may lead to chronic impairment of pulmonary function in ARDS survivors. 50 The amount of alveolar epithelial damage and impaired AFC capability are associated with impaired gas exchange and higher mortality.41,42,62 Injury of the alveolar epithelium (not the vascular endothelium) determines the progression to pulmonary fibrosis.42,67,68 Currently there is no specific treatment for post-ARDS pulmonary fibrosis other than supportive therapy.50,69
ARDS is one of the leading causes of death in ICU patients.38,40,41 Current ARDS therapy mainly constitutes supportive treatments, such as mechanical ventilation.51,70,71 This is predominantly effective in less severe cases, and may have serious side-effects.38,70,71 High tidal volume mechanical ventilation upregulates ACE expression and Ang-II activity, 71 activates JNK and ERK1/2, 72 and increases pulmonary parenchymal IL-6 levels via excessive alveolar distention. 70 Mechanical ventilation may promote ventilator-induced ALI, which is characterized by inflammation, increased vascular permeability, interstitial pulmonary edema, parenchymal infiltration, fibrosis, and thrombosis.42,48,71
The COVID-19-related ARDS may present atypically, in the sense that there is relatively well-preserved pulmonary compliance (despite the severity of hypoxemia), and systemic features of a hypercoagulable state.73–75 To understand how and why, a trinity of interconnected systems will be discussed, starting with the RAAS.
Renin angiotensin aldosterone system
To explain why changes in ACE2 and Ang-II levels are important in COVID-19, we need to discuss their interactions with other relevant systems, as well as their relation to comorbidities and complications.
If we exclusively focus on the RAAS, it is best described as a regulatory system with two axes that control vasoconstriction and vasodilation,3,76,77 which play an essential role in maintaining hemodynamic homeostasis.64,78,79 The classical ACE/Ang-II/AT1R axis promotes vasoconstriction.37,80,81 Renin increases Ang-I, which is subsequently converted to Ang-II by ACE,35,77,82 after which Ang-II exerts its cellular effects (predominantly via the AT1R).58,79,83 The counterregulatory ACE2/Ang-(1-7)/MasR axis promotes vasodilation.3,76,77 ACE2 converts Ang-II to Ang-(1-7), which exerts its cellular effects predominantly via the MasR.64,79,80
In a perfectly balanced RAAS, neither ACE nor ACE2 should be considered good or bad, as they are both required to maintain healthy homeostasis. Since ACE is required for either axis, Ang-II and ACE2 should be considered to be the main effectors of the RAAS. This balance can go either way, meaning low ACE2/high Ang-II or high ACE2/low Ang-II. Since only low ACE2/high Ang-II is relevant regarding COVID-19, the focus will be on that type of RAAS imbalance in particular.
Ang-II will be considered first, after which ACE2 will be discussed.
Angiotensin-II
Ang-II is considered to be the major player in the ACE/Ang-II/AT1R axis.41,83,84 A dysregulated RAAS (and associated elevated Ang-II levels) has many detrimental effects. Ang-II upregulates Aldosterone production and promotes hypertension,83–85 CVD,78,79,82 and fibrosis.71,80,86 Furthermore, elevated Ang-II levels increase PKC-mediated87–89 ROS production via NADPH oxidase86,90,91 and ΔΨM depolarization (through PKC-mediated modulation of KATP channels).58,91,92 Ang-II-mediated ROS production stimulates ERK1/2,58,88,93 JNK,91,94,95 and p38-MAPK,83,90,96 subsequently initiating crosstalk with NF-κB64,71,89 (Figure 1).

ROS- and IL-6-mediated MAPK signaling and their effects regarding severe COVID-19 complications, for example, ARDS and thrombosis.
ERK1/2, JNK and p38-MAPK are members of the MAPK family, and are preferentially activated by inflammation and environmental stresses (JNK and p38-MAPK in particular).97–99 MAPK signaling plays a crucial role in regulating cell apoptosis, inflammatory responses, and cell-cell junction formation.14,72,96 NF-κB is a transcription factor family that plays an important immunoregulatory role,44,100,101 and modulates the production of inflammatory cytokines44,86,89 and NADPH oxidase subunits.102–104
Ang-II-induced ROS production effectively induces insulin resistance,79,86,94 and exacerbates T2DM.64,86,89 Insulin resistance impairs the PI3K/Akt/eNOS pathway,94,105,106 subsequently reducing glucose uptake and NO production.86,88,89 Insulin signaling, and the role of Ang-II in insulin resistance, will be further discussed in the T2DM section. To understand the importance of Ang-II-mediated reduction of NO bioavailability in COVID-19, we will first consider the vascular endothelium.
Vascular permeability
The inner surface of the vascular tree is lined with a continuous monolayer of endothelial cells (joined together by tight-junctions), forming a protective selective permeability barrier between the circulating blood and the extravascular tissue.39,107,108 The endothelium is a metabolically active homeostatic organ, regulating the tone, structure and permeability of the vascular system in response to different stimuli (e.g. shear stress, ACh, and insulin).46,78,104 Limited vascular permeability is a function of a balanced endothelial phenotype, which constitutes smooth muscle relaxation, as well as low platelet activation and low fibrin formation.78,109,110 Endothelial dysfunction disrupts this balance and predisposes the vascular wall to inflammation, platelet activation, dysregulated coagulation, thrombosis, and increased vascular permeability.39,48,111
Both the disruption of endothelial tight-junctions39,72,108 and impaired NO bioavailability104,109,112 are two important causes of endothelial dysfunction and increased vascular permeability. ROS-mediated MAPK signaling is responsible for disruption of endothelial tight-junctions.39,72,108 Impaired NO bioavailability occurs via eNOS uncoupling,104,109,113 reduced eNOS activity,74,112,114 or ROS-scavenging.55,104,115 Prolonged and excessive vascular permeability can result in tissue damage, organ dysfunction, or even death.116,117
Impaired NO bioavailability plays a role in thrombosis, due to the hampering effect of NO on ROS-mediated upregulation of platelet activation, suggesting that either decreased NO bioavailability or increased ROS production is able to induce thrombosis.114,118 Endothelial dysfunction initiates the coagulation pathway by activating platelets and pro-coagulant cascades, while reducing anti-coagulant components and fibrinolysis.42,114 This results in pulmonary capillary microthrombi and fibrin deposition in parenchymal and interstitial compartments, 42 which is characterized by observed high D-dimer and von Willebrand Factor in some COVID-19 patients.21,119,120
During an inflammatory state, endothelial cells release von Willebrand Factor,110,120 which represents an important thrombotic risk factor.48,121 Von Willebrand Factor is a large adhesive glycoprotein, synthesized by endothelial cells,46,110,120 and is critical for platelet adhesion and aggregation.48,121 ABO blood group genes affect von Willebrand Factor expression, as well as their susceptibility to proteolytic degradation via ADAMTS13. 122 This is a possible explanation why ABO blood group type may be differentially related to COVID-19 severity. 122
Ang-II-mediated ROS production58,90,91 and decreased NO bioavailability79,94,123 promote vascular permeability41,81,83 and thrombosis6,80,82 (Figure 1). Ang-II-mediated disruption of ENaCs impairs AFC,41,64,124 exacerbating pulmonary edema.40,62,63 These are essential components of ARDS,41,42,62 which is a severe COVID-19 complication.25,76,125 Ang-II is significantly elevated in COVID-19 patients and is highly associated with viral load and lung injury.21,76,125
Angiotensin-converting enzyme 2
The ACE2/Ang-(1-7)/MasR axis can mitigate Ang-II-mediated negative effects.77,79,81
ACE2 is a homologue of ACE,78,82,84 and a key component of the RAAS.77,81,126 ACE2 is a two-part type-I transmembrane protein, consisting of a glycosylated extracellular N-terminal domain (containing the SARS-CoV-2-binding carboxypeptidase site), and an intracellular C-terminal cytoplasmic tail.3,127,128 The extracellular catalytic domain of ACE2 can be cleaved and released by ADAM-175,6,76 or TMPRSS2.2,3,8 ACE2 is widely expressed in the heart, kidneys, lungs, CNS, and intestines,80,82,84 and is present in type-II pneumocytes and endothelial cells.6,71,129 ACE2 is predominantly membrane-bound, although it does exist, with a short half-life,52,130 in soluble form.3,6,127
Increased ACE2 expression, upregulating the ACE2/Ang-(1-7)/MasR axis, has multiple beneficial effects, for example, mitigation of hypertension,80,84,131 CVD,77,82,129 insulin resistance,79,132,133 and T2DM.5,25,134 ACE2 also increases NO bioavailability,79,80,135 subsequently mitigating endothelial dysfunction,76,82,133 vascular permeability43,64,76 and thrombosis.6,76,136 Furthermore, via decreased MAPK signaling,44,71,137 ACE2 reduces inflammation,3,43,71 ROS production,78,138,139 and neutrophil accumulation.56,76,95 In this manner, ACE2 protects against pulmonary edema,35,75,76 fibrosis,64,71,77 and ARDS.38,43,81
Low ACE2 expression has been associated with hypertension, CVD, T2DM, inflammation, and ARDS,4–6 which happen to be risk factors for (severe) COVID-19 complications.1,15,23 Since ACE2 is the SARS-CoV-2 cell-entry point,2,3,6 there has been speculation that elevated ACE2 expression may increase susceptibility for SARS-CoV-2 infection.140–142 ACE2 expression is higher in females than in males,4,134 and declines with age5,141 (in men more so than in women).6,143 This can be explained by the upregulation of ACE2 expression by Estrogen,4,131 and the X-chromosomal location of the ACE2 gene3,129,144 (which is regulated epigenetically via DNA methylation).12,13 DNA methylation is associated with biological age,12,145,146 which suggests that biological age may be a more accurate risk factor for severe COVID-19 complications compared to chronological age.13,147 This pattern of ACE2 expression may partly explain why elevated ACE2 levels possibly have no negative effects on COVID-19 susceptibility.4,6,134 Also, there is a negative correlation between ACE2 expression and COVID-19 fatality.4,5,144 SARS-CoV-2-induced downregulation of ACE2 expression may especially be detrimental in people with comorbidities, since they initially have a lower ACE2 baseline.4,6,21 Additional COVID-19-mediated ACE2 deficiency may amplify the RAAS dysregulation,5,6,76 resulting in upregulation of Ang-II, which is indeed significantly elevated in COVID-19 patients.21,76,125 In the lungs, such dysregulation can induce the progression of inflammatory and thrombotic processes, because Ang-II is now unopposed by ACE2.6,21,125 This suggests that ACE2 may not be the culprit in COVID-19, but may actually have an important protective role, despite the ACE2-mediated cell-entry mechanism of SARS-CoV-2.5,76,134 A low ACE2 expression (or reserve) may contribute to the progression of COVID-19 to a (more) severe or fatal stage.4,6,125 Because of the high intrinsic affinity of ACE2 with the SARS-CoV-2 spike (S1) proteins, 6 a lower ACE2 expression may not affect the susceptibility for SARS-CoV-2 infection at all.6,127
Although many effects of ACE2 have been attributed to the downregulation of Ang-II levels, other substrates play a major role in ACE2-related functions as well.56,64,81
The framework we have now established consists of the following hypothesis: “a SARS-CoV-2-induced RAAS imbalance, comprised of reduced ACE2 expression (and a subsequent elevated Ang-II activity), plays a role in severe COVID-19 complications.”
There are still many loose ends in this narrative. In order to connect these, two more systems will be discussed, that is, the CS and the KKS. The CS and the KKS are linked.107,148,149 The KKS and the RAAS are linked as well.25,56,64 The RAAS, CS, and KKS, form a trinity of systems with several regulatory axes contributing to severe COVID-19 complications.
Complement system
The CS is an important component of the innate immune system, involved in host defense against micro-organisms, clearance of immune complexes and removal of apoptotic cells,149–151 and is intrinsically linked to the coagulation pathway.21,151,152 The CS is a key mediator of lung damage during (corona virus) infections, raising the possibility that CS activation may play a role in severe COVID-19 complications.16,74,153
Although most CS components are synthesized in the liver, type-II pneumocytes provide local CS proteins.154–156 The CS components C1, C3a, C5a, as well as the C5b-C9 MAC, all contribute to increased endothelial permeability.107,157,158
The CS can be activated via three pathways, that is, the classical, lectin, or alternative pathway.154,159,160 The classical pathway entails the creation of immune complexes with IgM/IgG antibodies, binding predominantly to pathogenic antigens.107,159,161 C1 binds to the antibody and forms C3 and C5, via initiating a series of enzymatic cascades.107,154,162 C3 and C5 are both cleaved to C3a/C3b and C5a/C5b respectively.107,154,160 C3a and C5a act as chemotactic agents for phagocytes.107,151,154 C3b is involved in opsonization of pathogens that are subsequently destroyed by phagocytes.107,154,159 C5b-C9 forms a MAC that induces cell-lysis by punching holes through their membranes.107,154,161 The lectin pathway involves the interaction of MBL with the pathogen (or the surface of a pathogen-infected cell), followed by the subsequent binding and activation of MASP2, directly activating similar CS cascades as in the classical pathway.74,153,154 The alternative pathway involves a spontaneous conformation change of C3, and after a short cascade, C3 is cleaved into C3a and C3b without the use of antibodies, leading to similar cascades as the classical pathway.151,154,163 The CS is inhibited by the SerPin C1INH via inhibition of C1 in the classical pathway, MASP2 in the lectin pathway, and C3b in the alternative pathway.107,149,164
Excessive CS activation can lead to inflammation and (excessive) neutrophil recruitment via opsonization and (via C3a- and C5a-mediated)107,153,156 chemotaxis.16,148,153 Excessive CS activation (on the endothelial surface) can lead to vascular problems, for example, endothelial damage (via MAC-induced lysis),74,148,154 vascular permeability,107,148,158 thrombosis,148,151,152 (diffuse) TMA,159,162,165 and DIC.161,166,167 This is how excessive CS activation exacerbates ARDS,16,74,154 pulmonary fibrosis,50,168,169 and possibly organ dysfunction.16,151,159
In at least a subset of severe COVID-19 patients an excessive CS activation is found via the lectin pathway,153,170,171 as demonstrated by elevated C5b-C9 MAC components, MBL-MASP2 in the pulmonary microvasculature, and parenchymal neutrophils. 74 This is consistent with sustained and systemic CS activation and an associated pro-coagulant state.21,74 A possible modus operandi for additional CS activation via the lectin pathway in COVID-19 is the binding of SARS-CoV-2 N-proteins to MASP2. 153 This leads to excessive CS activity,153,171 further exacerbating CS-mediated MAC formation, inflammation, and concurrent activation of the coagulation pathway,21,74 resulting in ARDS16,153 and thrombosis74,170 (Figure 2). The observed high D-dimer and von Willebrand Factor (and its associated thrombotic activity) in severe COVID-19 patients,111,119,120 can at least be partially explained by excessive CS activation (and associated MAC-mediated endothelial dysfunction), followed by an activated coagulation pathway.74,153,159
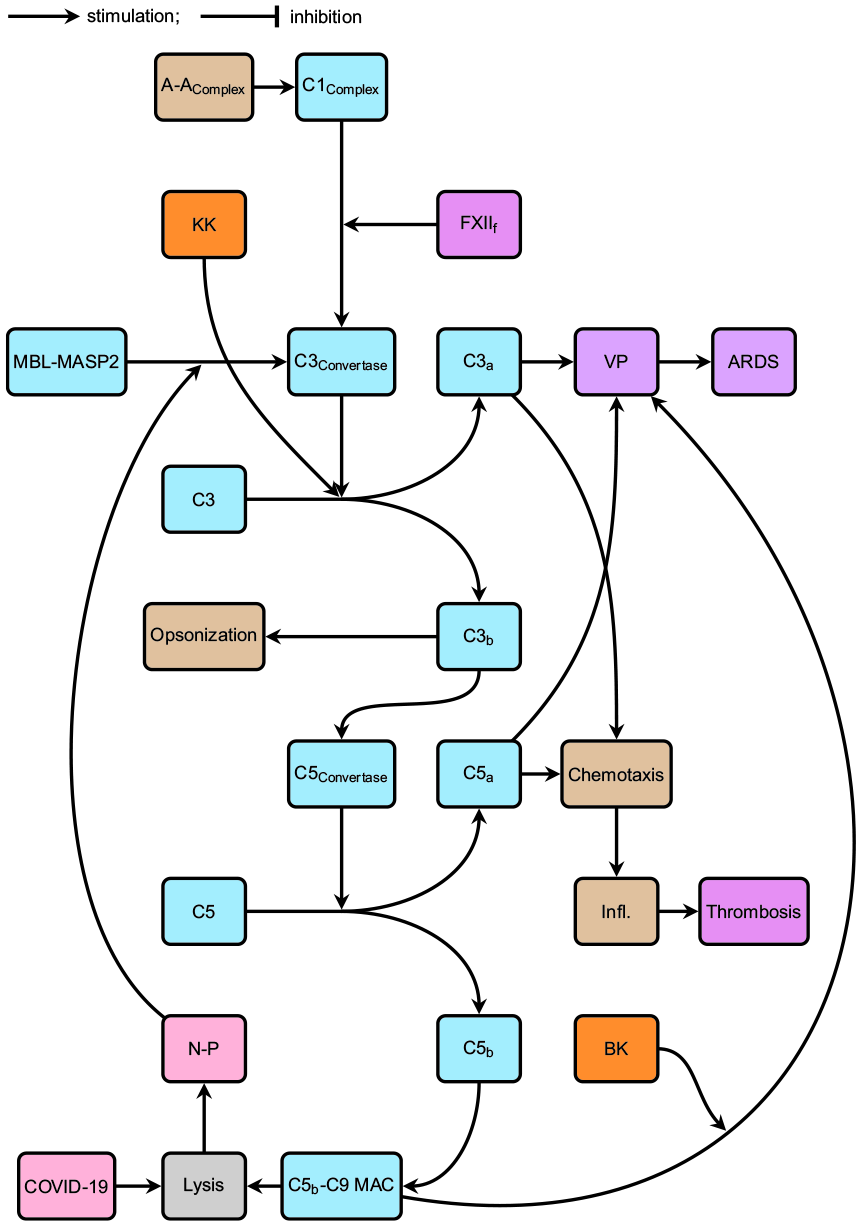
The CS including the COVID-19-induced lectin pathway feedback mechanism.
Our framework can now be expanded with the hypothesis: “an excessive CS activation plays a significant role in severe COVID-19 complications.”
The CS is not only linked to the coagulation pathway,151,161,166 but to the KKS as well,107,148,149 which will be discussed next.
Kallikrein-kinin system
The KKS, like the CS, is an important part of the innate immune system,149,172,173 and is activated during inflammation.148,164,174 The KKS consists of Hageman Factor (Factor XII), PK, HK, the proteolytic KK enzymes, and effector peptides, such as BK (and its active metabolite des-Arg9-BK).107,149,164 Factor XII gets activated via inflammatory processes and converts to Factor XIIa, which in turn converts PK into KK, initiating the kinin cascade107,149,164 (cleaving HK to generate BK and other metabolites, such as des-Arg9-BK).85,148,164 Factor XIIa also turns Factor XI into Factor XIa, subsequently activating the coagulation pathway, linking it to the KKS.107,149,175 KK turns Factor XIIa into Factor XIIf, which initiates the CS via C1r cleavage, linking the KKS directly to the CS.107,149,176 KK also turns pro-Renin into Renin, linking the KKS directly to the RAAS.64,177,178 All these processes are inhibited by the SerPin C1INH.107,149,164
The kinin BK is not very stable, and is easily degraded by ACE.81,85,88 Another kinin, des-Arg9-BK, is much more stable than BK,148,164,179 but can be degraded by ACE2.25,56,64 This shows another direct link between the KKS and the RAAS.36,75,81
KKS signaling is mediated by two receptors, B2R and B1R.85,148,180 While the B2R is ubiquitously expressed in most healthy tissues, B1R is synthesized de-novo and upregulated as a consequence of tissue injury or inflammatory processes.81,164,180 BK is the primary ligand for B2R, whereas des-Arg9-BK is the primary ligand for B1R.85,107,148
Kinins are potent inflammatory mediators,36,88,164 increasing vascular permeability85,180,181 and neutrophil recruitment.56,148,174 This may lead to inflammation,25,148,180 edema,75,164,180 and pain.85,164,174 Uncontrolled and excessive KKS activation, or abnormal kinin degradation, can cause an acute accumulation of BK and/or des-Arg9-BK, leading to excessive inflammation.56,107,148 Excessive KKS activation may also promote T2DM via (MAPK-mediated)181–183 destruction of pancreatic Langerhans islets,85,184,185 and increase vascular permeability.148,164,180 This results in exacerbated edema,85,107,164 neutrophil migration,56,148,174 thrombosis,148,186,187 ARDS,25,75,188 and possibly even organ dysfunction.25,187,189
The KKS and the CS are intrinsically linked at multiple levels, and are both activated during (vascular) inflammation (i.e. via gC1qR, which binds both C1q and HK).107,148,149 Simultaneous and uncontrolled excessive activation of both the KKS and CS (on the endothelial surface) is largely responsible for increased vascular permeability and edema.107,148,149 KK, which cleaves HK and subsequently releases BK, also cleaves and activates C3.148,190,191 The KKS107,148,149 and CS150–152 are both intrinsically linked to the coagulation pathway. The endothelial permeability-inducing effect of the C5b-C9 MAC is regulated by BK.107,192,193 The KKS and CS are both inhibited by C1INH.148,149,164
ACE2 degrades the otherwise stable des-Arg9-BK.56,64,81 SARS-CoV-2 reduces ACE2 expression.3,6,125 A reduction in pulmonary ACE2 subsequently increases Ang-II5,125,194 and impairs the degradation of the des-Arg9-BK/B1R axis of the KKS.6,25,56 This exacerbates neutrophil migration56,148,174 and ARDS.25,56,75 The des-Arg9-BK/B1R axis of the KKS is not affected by corticosteroids,195–197 which means that as long as the virus is present, ACE2 will not be, and the kinin-induced ARDS will persist. 75
We now have established multiple connections between the RAAS, CS, KKS, and the coagulation pathway. In our trinity of systems, the RAAS3,76,77 controls vasoconstriction and vasodilation, whereas the KKS85,180,181 and the CS107,158,198 control vascular permeability and vasodilation. ACE2 is the one ring that rules them all 75 (Figure 3). The SerPin C1INH effectively suppresses the KKS,107,149,164 the coagulation pathway,107,151,152 and all three pathways of the CS.150,152,176

The trinity of systems: RAAS, CS, KKS, and their interconnections, as well as their interactions with IR, the coagulation pathway, inflammation, and COVID-19.
Our framework can now be further expanded with the hypothesis: “a SARS-CoV-2-induced reduction of ACE2 expression directly causes a disruption and excessive activation of the KKS, which is not only able to further activate an already active CS, but directly plays a significant role in the exacerbation of severe COVID-19 complications.”
Next, the T2DM interactions with our trinity of systems will be discussed.
Type-2 diabetes mellitus
T2DM is considered to be a metabolic disease, comprising insulin resistance and pancreatic β-cell dysfunction, resulting in insulin deficiency and subsequent hyperglycemia.99,104,199 T2DM is characterized by comorbid conditions of CVD89,104,200 and hypertension.89,201,202 T2DM is also associated with vascular problems, for example, endothelial dysfunction, vascular permeability, platelet dysfunction, and hypercoagulation.113,203,204 This can be partially contributed to a dysregulated RAAS5,94,105 (low ACE24,5,82 and high Ang-II79,94,106). CVD is the main complication of T2DM.114,204,205 However, clinical CVD can also precede the development of T2DM,200,206,207 suggesting that T2DM and CVD may both have an underlying cause, for example, a chronic low-grade inflammatory state,199,208,209 insulin resistance,53,210,211 or a dysregulated RAAS.86,89,133
Obesity
In T2DM, macrophage infiltration into expanding adipose tissue and pancreatic islets,212–214 as well as macrophage polarization toward the M1 phenotype99,215,216 (as a result of IL-6 trans signaling),53,214,217 is involved in the development of chronic low-grade inflammation in obese individuals.199,208,218 Visceral adipose tissue in particular, is characterized by high secretion of inflammatory cytokines, and T2DM patients have more visceral adipose tissue than nondiabetics.53,98,219 Furthermore, inflammatory cytokines in adipose tissue stimulate JNK and NF-κB,99,214,220 which induces insulin resistance.98,199,221 Finally, the RAAS may be disrupted in expanding visceral adipose tissue, exacerbating inflammation and insulin resistance.6,222,223
Insulin resistance
P(Tyr)IRS-1 is required for insulin-stimulated activation of the PI3K/Akt/eNOS pathway99,206,224 and multiple downstream effectors that promote glucose uptake and NO production.112,225,226 NO has vasoprotective effects,79,227,228 and its bioavailability depends on the balance between the rate of its eNOS-mediated production and its ROS-mediated inactivation.104,109,229
Increased P(Ser)IRS-1 and decreased P(Tyr)IRS-198,224,230 (both induced by JNK and ERK1/2) disrupt insulin signaling,94,199,206 preventing activation of the PI3K/Akt/eNOS pathway.99,115,231 Chronic P(Ser)IRS-1 also targets IRS-1 for degradation or migration to inaccessible subcellular compartments.54,89,232
Hyperinsulinemia, caused by insulin resistance, activates both JNK230,232,233 and ERK1/2.88,234,235 Subsequently this further stimulates P(Ser)IRS-1,97,99,230 which is how hyperinsulinemia exacerbates insulin resistance.94,232,236 Hyperinsulinemia also upregulates Ang-II, which is how insulin resistance may disrupt the RAAS.89,237,238
Ang-II-mediated ROS production increases P(Ser)IRS-179,88,94 and decreases P(Tyr)IRS-1,86,89,105 effectively inducing insulin resistance87,239,240 (Figure 1). A vicious cycle between the RAAS and insulin resistance has now been created.89,241,242
Insulin resistance is a risk factor for T2DM,86,206,232 obesity,85,98,106 hypertension,88,89,105 and CVD.87,210,243
β-Cell dysfunction
The progression from insulin resistance to T2DM implicates the inability of pancreatic β-cells to compensate for increased insulin demand.53,221,244
Sustained JNK activation causes pancreatic β-cell dysfunction,230,232 especially in obese individuals with an exacerbated inflammatory milieu.99,199,221
IL-6 increases the proliferation of α-cells and apoptosis of β-cells in the pancreas,53,245,246 suggesting a link between IL-6 and T2DM.208,218,247 The role of IL-6 in the proliferation of α-cells may initially compensate for the impaired β-cells in T2DM and contributes to limit hyperglycemia.53,245,248
Hyperglycemia increases IL-6 levels, both systemically and locally in pancreatic Langerhans islets,53,249,250 and promotes β-cell death99,246,251 (Figure 1).
In T2DM, the KKS is activated and kinins cause damage to pancreatic Langerhans islets, suggesting a link between the KKS and T2DM. 85
Furthermore, in T2DM, ACE2 expression is downregulated,15,133 and increased Ang-II plays a part in reducing β-cell function via ROS-mediated apoptosis,89,252 suggesting a link between RAAS disruption and T2DM.64,86,89
Hyperglycemia
Pancreatic β-cell dysfunction leading to T2DM results in insulin deficiency and subsequent hyperglycemia,104,115,221 which has a plethora of detrimental effects.
Hyperglycemia increases the production of IL-653,55,250 and AGEs.104,113,253 AGEs bind to RAGEs99,104,254 (which are expressed on the surface of many cell types, including endothelial, epithelial, and immune cells),62,113,255 and subsequently increases PKC-mediated55,256,257 ROS production via NADPH oxidase.118,203,258 Hyperglycemia-mediated ROS production stimulates ERK1/2,259–261 JNK,99,262,263 and p38-MAPK,264–266 subsequently initiating crosstalk with NF-κB86,104,267 (Figure 1). This ROS-mediated MAPK signaling downregulates ACE2,14,15,268 and upregulates ACE,14,268,269 Ang-II,89,268,270 ADAM-17,5,271,272 and IL-6.114,249,267 It also disrupts endothelial tight-junctions,45,108,257 and reduces NO bioavailability (via eNOS uncoupling,113,273,274 decreased NO production,55,115,267 and NO scavenging104,109,275). Hyperglycemia-mediated ROS production effectively promotes endothelial dysfunction.104,108,113 Therefore hyperglycemia increases vascular permeability,39,115,276 and subsequently promotes (pulmonary) edema.55,203,277
Hyperglycemia also promotes thrombosis,24,113,118 via platelet activation, increased coagulation factors, and decreased fibrinolysis.114,205,278
Furthermore, hyperglycemia (even short-term) dysregulates both the innate and adaptive immune system.24,55,265 This may affect neutrophil-IL-6 signaling, chemotaxis, phagocytosis, respiratory burst, anti-microbial activity, and production of inflammatory cytokines.23,55,266 This creates a milieu in which SARS-CoV-2 can flourish.15,23,24
Finally, hyperglycemia also interferes with the CS, upregulating expression of several CS component genes, hindering C3b-mediated opsonization and IG-function via glycation.55,279,280
Well-controlled blood-glucose levels reduce the risk of severe COVID-19 complications.23,24,281 Therefore, our framework can now be further expanded with the hypothesis: “T2DM-induced hyperglycemia is not only just a risk factor for severe COVID-19 complications, but is actually exacerbated (possibly even induced) by COVID-19.”
Next, the role of IL-6 signaling, and the interactions with T2DM and COVID-19 complications, will be discussed.
Interleukin 6
IL-6 has both pro- and anti-inflammatory characteristics.53,208,282 Dysregulated or excessive IL-6 signaling is considered to be involved in insulin resistance,104,206,247 β-cell dysfunction,53,245,251 T2DM,53,208,218 and CVD.208,283,284
Various cell types can locally produce IL-6,284–286 which can be transported through the circulation, affecting more distant regions in the body. 53 Immune cells are simultaneously both sources and targets of IL-6.53,283,286 Beta-coronavirus infection of immune cells, for example, monocytes, macrophages, and dendritic cells, will result in their activation and subsequent secretion of IL-6. 287
To exert its physiological effects, IL-6 utilizes both the classic and trans signaling pathway.208,213,288 In classic signaling, IL-6 binds to mIL-6R and forms a complex with gp130, after which downstream signaling is mediated via JAK and STAT3.283,284,286 In trans signaling, extracellular sIL-6R can bind to IL-6, forming a complex with gp130, after which downstream signaling is mediated via JAK and STAT3 in cells that do not express mIL-6R53,284,289 (such as endothelial53,116,284 and pancreatic α and β cells245,246,248). IL-6 classic signaling is mostly involved in anti-inflammatory activities, whereas IL-6 trans signaling is mostly involved in pro-inflammatory activities.53,213,289
Most cells express gp130, while mIL-6R is mostly found on monocytes, macrophages, neutrophils, and α- and β-cells of pancreatic Langerhans islets.53,208,245 The widespread expression of gp130 on most cell types, including endothelial cells, dramatically expands the range of IL-6 target cells using trans signaling.208,284,287 In order to prevent a systemic response to IL-6 trans signaling, sgp130 specifically blocks trans signaling without affecting classical signaling, and basically constitutes a physiological buffer for circulating IL-6.53,284,288
IL-6 activates ERK1/2,54,206,290 JNK,291–293 and p38-MAPK,89,294,295 subsequently initiating crosstalk with NF-κB44,104,249 (Figure 1). This IL-6-mediated MAPK signaling decreases P(Tyr)IRS-154,104,247 and increases P(Ser)IRS-1,206,296,297 inducing insulin resistance53,89,296 and impairing NO production.54,206,290 IL-6-mediated MAPK signaling also disrupts endothelial tight-junctions116,285,298 and induces endothelial cell contraction.285,299 Therefore, IL-6 effectively increases vascular permeability70,104,285 and exacerbates edema 116 and lung injury/ARDS.70,300,301 The lack of mIL-6R on endothelial cells indicates trans signaling as the main mechanism involved in the detrimental effects of IL-6 on vasculature.53,116,284
IL-6 also induces epigenetic changes (regarding genes involved in insulin signaling) via DNMT-induced alteration of DNA methylation patterns, promoting endothelial dysfunction54,302 and insulin resistance.213,303,304
IL-6 levels are significantly elevated in COVID-19 patients.22,305,306 IL-6 plays an important role in CRS,16,306,307 exacerbates ARDS,70,287,305 and predicts mortality282,305,308 (Figure 4).
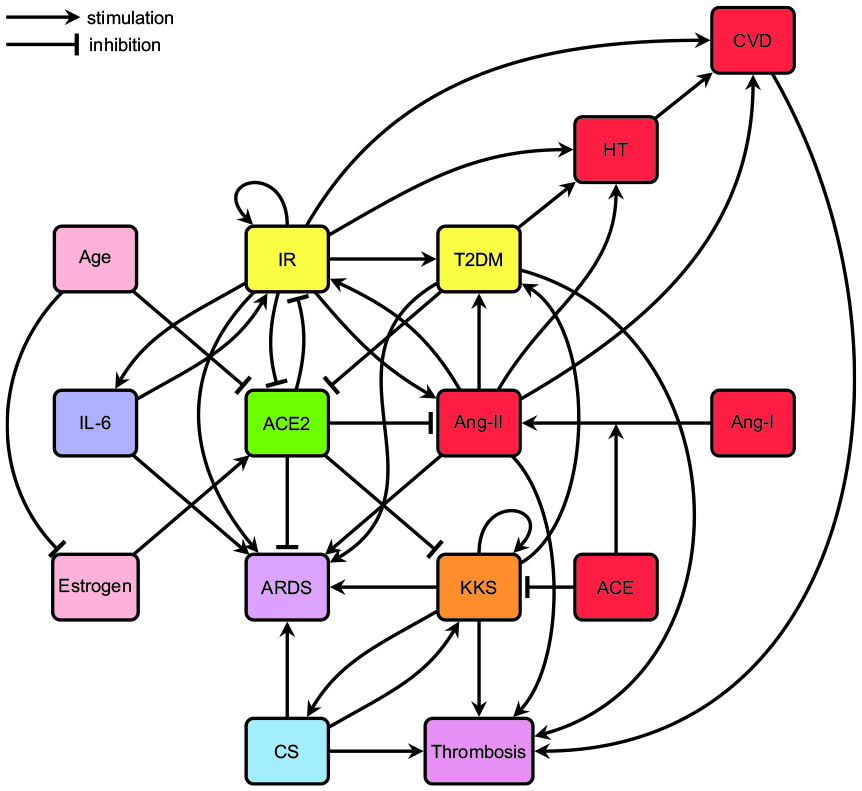
The RAAS, CS, KKS, IR, IL-6, and their interactions regarding severe COVID-19 complications, for example, ARDS and thrombosis, with ACE2 as master regulator.
Our framework can now be further expanded with the hypothesis: “Excessive IL-6 trans signaling significantly contributes to severe COVID-19 complications.”
In the last section, we will briefly discuss some potential attractive therapeutic targets and their pharmacological interactions.
Pharmacological interactions
Due to the lack of approved targeted medication for COVID-19,16,22,309 and developing an effective and safe vaccine may require some time, it is recommended to explore multiple potential therapeutic targets to mitigate severe COVID-19 complications.
TMPRSS2 inhibition
Since TMPRSS2 is essential for SARS-CoV-2 membrane fusion and subsequent cell-entry,2,3,7 inhibition of TMPRSS2 activity can potentially prevent severe COVID-19 complications.3,8,310 Nafamostat Mesylate, a clinically approved and safe medication for pancreatitis, inhibits TMPRSS2-mediated SARS-CoV-2 envelope-membrane fusion and subsequent cell-entry.3,311,312 Nafamostat Mesylate may also protect against thrombosis and DIC via its anti-coagulant properties.3,313
ACE inhibition/angiotensin II receptor blockage
Both ACE-inhibitors and ARBs reduce hypertension,85,96,129 CVD,80,96,129 pneumonia,314–316 insulin resistance (and T2DM).85,86,106 They also increase ACE2 expression1,76,80 and mitigate pulmonary edema.71,317,318 ACE-inhibitors however, also inhibit BK degradation, subsequently increasing BK, possibly increasing vascular permeability75,107,149 and systemic acquired angioedema.75,149,319
Because ACE-inhibitors and ARBs upregulate ACE2 expression,1,76,80 and SARS-CoV-2 uses ACE2 for cell-entry,3,6,8 it has been speculated that ACE-inhibitors and ARBs can have negative effects in COVID-19 patients.140–142 Multiple studies however, show no association between both ACE-inhibitors and ARBs, and increased susceptibility for COVID-19 or the severity of its complications.320–322
Various experimental models37,71,76 show that rhsACE2 activates the protective ACE2/Ang-(1-7)/MasR-axis of the RAAS,71,76,80 degrades des-Arg9-BK, 75 and improves ARDS symptoms.44,71,82 Therapeutic use of rhsACE2 in COVID-19 patients may have beneficial effects, such as effectively sequestering circulating viral particles to prevent S-protein interactions with membrane-bound ACE2, while simultaneously rebalancing the RAAS into a more protective equilibrium.6,76,323 Clinical-grade rhsACE2 can significantly inhibit SARS-CoV-2 infections in vitro (in a dose-dependent manner), reduce viral load by a factor of 1.000 – 5.000, 324 and is currently being tested in a phase-II trial in Europe.5,127
Complement/kinin inhibition
Inhibiting an excessively active CS may reduce severe COVID-19 complications, for example, ARDS, 153 pulmonary fibrosis,50,154 (diffuse) TMA,159,171 and DIC.74,151,153 Eculizumab is a monoclonal antibody that binds to C5 and prevents cleavage into C5a and C5b, as well as the formation of the C5b-C9 MAC,16,74,325 and may also reduce the risk of TMA162,163,165 and DIC.166,326
Inhibiting the KKS, either by inhibiting kinin production or blocking B1R/B2R, may improve COVID-19-induced ARDS.25,75,327 Since B1R is upregulated during inflammation,56,107,180 and des-Arg9-BK is much more stable than BK,148,164 the des-Arg9-BK/B1R axis is of major importance in vascular permeability during inflammation. 164 Unfortunately, there currently is no approved B1R-inhibitor available.25,75,327 Icatibant is a selective B2R antagonist, traditionally used for hereditary angioedema,25,107,164 and may improve severe COVID-19 complications.75,327 B2R inhibition, besides reducing the KKS, also reduces the CS. 148
C1INH inhibits the activation of both the CS and KKS, 148 qualifying it as an attractive potential therapeutic option for severe COVID-19 complications. Plasma-derived C1INH, a first-line therapy for hereditary angioedema, appears to be safe. 328 Ruconest, a recombinant human C1-esterase-inhibitor, has achieved some good preliminary results in treating COVID-19 patients, according to a press release. 329
Interleukin 6 inhibition
IL-6 inhibition mitigates vascular permeability and alveolar-capillary barrier disruption.70,116,309
Tocilizumab is a monoclonal antibody, mainly used for the treatment of rheumatoid arthritis.22,308,330 It inhibits IL-6305,306,331 via binding to both mIL-6R and sIL-6R, resulting in complete blockade of both classic and trans IL-6 signaling pathways.287,308,330 Disruption of both pro- and anti-inflammatory activities of IL-6 may cause side-effects, for example, secondary (bacterial or fungal) infections, liver malfunction, or hypercholesterolemia.53,208 Despite the possible side-effects, preliminary studies show that Tocilizumab appears to be effective and safe regarding severe COVID-19 complications.22,306,309 Tocilizumab however, seems less effective in hyperglycemic patients. 332
Sgp130Fc (a recombinant version of sgp130) specifically blocks IL-6 trans signaling, without affecting the anti-inflammatory and protective classical IL-6 signaling.53,208,288 Therefore sgp130Fc may result in better therapeutic outcomes with fewer undesired side-effects.53,208,333 Sgp130Fc has demonstrated robust efficacy in the treatment of several autoimmune and inflammatory conditions53,288 with fewer side-effects than global IL-6-inhibitors.53,208,333 Sgp130Fc may potentially have therapeutic benefits for COVID-19-induced ARDS. 333
Summary
We have established the concluding framework regarding the COVID-19 pathophysiological crossroads: “COVID-19 disrupts the RAAS balance via reduction of ACE2 expression and a subsequent elevation of Ang-II. COVID-19-induced reduction of ACE2 expression induces excessive activation of the KKS and subsequently the CS. T2DM-induced hyperglycemia is not only just a risk factor for severe COVID-19 complications, but is actually exacerbated (possibly even induced) by COVID-19. Ang-II, excessive KKS and CS activation, hyperglycemia, and IL-6 trans signaling, all contribute significantly to severe COVID-19 complications (e.g. ARDS and thrombosis/DIC).” The RAAS, CS, KKS, and the coagulation pathway, are all intrinsically connected sensitive systems, with ACE2 as master regulator. Elevated ACE2 baseline levels may protect from severe COVID-19 complications.
There are no effective approved medications against COVID-19 yet, and a vaccine may be a long way off. However, several attractive therapeutic targets in the RAAS, CS, and KKS have shown promising preliminary results against severe COVID-19 complications.
Footnotes
Appendix
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.