
Abstract
Hypothesis/Introduction:
The pathophysiological role of oxidative stress (OxSt) in hypertension and target organ damage is recognized. Angiotensin II (Ang II) induces OxSt via NAD(P)H oxidase activation and production of proinflammatory cytokines/growth factors leading to cardiovascular-renal remodeling. Ang II stimulates the RhoA/Rho kinase (ROCK) pathway, which is deeply involved in the development of cardiovascular-renal remodeling via OxSt induction. Olmesartan, an Ang II type 1 receptor blocker, possesses antioxidant and activating nitric oxide system-related effects, which we have shown in terms of p22phox reduction, heme oxygenase-1 and calcitonin gene-related peptide increase. This study evaluates in 15 untreated hypertensive patients the effect of olmesartan treatment on p63RhoGEF, key in Ang II-induced ROCK activation, and MYPT-1 phosphorylation, a marker of ROCK activity.
Materials and methods:
The p63RhoGEF protein level and MYPT-1 phosphorylation (Western blot) were evaluated at baseline, and after three and six months of olmesartan treatment.
Results:
Olmesartan normalized systolic and diastolic BP (p < 0.001), reduced p63RhoGEF level: 1.3±0.25 d.u. (baseline) vs 1.0±0.29 (three months), p < 0.0001 vs 1.0±0.22, (six months), p < 0.0001 and MYPT-1 phosphorylation: 1.2 ±0.14 (baseline) vs 0.9±0.19 (three months), p = 0.008, vs 0.8±0.16 (six months), p = 0.001.
Conclusions:
These data added to our previous results further provide a mechanistic rationale for olmesartan’s antioxidant/anti-inflammatory potential translation, in the long term, toward anti-atherosclerotic/anti-remodeling effects reported by clinical trials.
Introduction
RhoA/Rho kinase signaling and its relationships with angiotensin II (Ang II), oxidative stress and nitric oxide (NO) level are deeply involved in cardiovascular biology and pathophysiology.1–3 The Ang II-mediated activation of this pathway following Ang II type 1 receptor (AT1R) stimulation, in fact, plays an essential role in both the increase of vascular tone and hypertension via Ca2+ sensitization of smooth muscle contraction 4 and increase of myosin light chain (MLC) phosphorylation through the Rho kinase-mediated inhibition of myosin phosphatase-1 (MYPT-1) activity, 2 and in the induction of long-term complications of hypertension such as changes in cardiovascular structure and induction of atherosclerosis,1,2 these latter essentially via induction of oxidative stress.
Guanine nucleotide exchange factors (RhoGEFs) activate Rho GTPases including RhoA by catalyzing the exchange of GDP to GTP,1,2 allowing the interaction of G proteins with downstream effectors resulting in the control of multiple vascular cell processes including contraction, proliferation and migration. p63RhoGEF has been reported as a specific mediator transducing the Ang II message from activated AT1R via Gq protein to RhoA/Rho kinase activation 5 via binding of the α subunit of Gq protein.6,7 This leads to effects of Ang II-mediated activation of RhoA/Rho kinase on the cardiovascular system, such as vascular contraction, proliferation and cardiovascular remodeling.1,2
The involvement of RhoA/Rho kinase in these processes is based on the following evidence: (i) the increased level of p63RhoGEF and increased MYPT-1 phosphorylation in hypertensive patients;8,9 (ii) the RhoA/Rho kinase activation of NAD(P)H oxidase, 10 essential for the induction of oxidative stress; (iii) the demonstration of RhoA/Rho kinase signaling involvement in the induction of atherothrombogenesis1,10 and (iv) the Rho kinase inhibition, which leads to cardiovascular protection and the prevention of atherogenesis.1,10
Among antihypertensive drugs, olmesartan, an AT1R blocker, widely used in the treatment of hypertension, has been shown to possess antioxidant and activating NO system-related effects. Consistent results with olmesartan and oxidative stress-related processes have been, in fact, obtained both in in-vitro and in animal models.11-14 We have recently reported in essential hypertensive patients that treatment with olmesartan reduced oxidative stress, the phosphorylation state of extracellular signal-regulated protein kinases 1 and 2 (ERK 1/2), the effector of oxidative stress-mediated Ang II signaling for cardiovascular remodeling and the plasma level of oxidized low-density lipoprotein (oxLDL), a marker of oxidative stress. 15 In addition, treatment of hypertensive patients with olmesartan has increased the level of heme oxygenase-1, 15 a potent antioxidant and anti-inflammatory protein,16,17 and the number of circulating endothelial progenitor cells (cEPCs), reduced cEPCs apoptosis and increased the level of calcitonin gene-related peptide, 18 a vasorelaxant that reverses Ang II-induced senescence of EPCs and reduces blood pressure (BP) in spontaneously hypertensive rats. 19
This study aims to evaluate in mononuclear cells from essential hypertensive patients the effect of a relatively short-term treatment with olmesartan on the RhoA/Rho kinase pathway. To this end we have evaluated the protein expression of p63RhoGEF, a guanine nucleotide exchange factor, key mediator of the RhoA/Rho kinase activation by Ang II,5–7 and the phosphorylation state of MYPT-1, a marker of Rho kinase.1,2
Patients and methods
Patients
The study was carried out on 15 uncomplicated, nonsmoking and never-before-treated essential hypertensive patients (10 men, five women, aged 35–62 years) attending our hypertension clinic, Department of Medicine, at the University of Padova, Italy. Their BP ranged from 147 to 157 mmHg systolic blood pressure (SBP) and 96 to 100 mmHg diastolic blood pressure (DBP).
Diabetes was ruled out by fasting serum glucose test less than 126 mg/dl and impaired renal function by serum creatinine less than 1.0 mg/dl and urinary albumin/creatinine excretion less than 30 mg/g. Secondary hypertension was excluded by the evaluation of plasma renin activity and plasma aldosterone before and after 50 mg of captopril (captopril test). Lipid profile was normal and patients were not taking lipid-lowering drugs or aspirin. Body mass index (BMI) was also normal (<25 kg/m2). None of the patients had cardiac failure, evidence of coronary heart disease or established left ventricular hypertrophy by conventional M-mode echocardiography.
Informed consent was obtained from all study participants.
After enrollment (baseline), patients were treated with olmesartan medoxomil (20 mg per day) and were seen after one, three and six months. BP was measured in the sitting position with a mercury sphygmomanometer, to the nearest 2 mmHg by a trained observer. The mean of three consecutive measurements, two minutes apart, was used for analysis of the results.
At baseline and at three and six months, 35 ml of peripheral blood was withdrawn from the antecubital vein after overnight fast for ex-vivo molecular biology determinations.
All patients followed a diet containing approximately 150 mmol sodium per day, and no substantial difference in the patients’ usual diet was revealed by a dietary questionnaire administered at three and six months, particularly regarding salt and alcohol consumption.
Methods
Mononuclear cell preparation
Peripheral blood mononuclear cells (PBMCs) from 35 ml of ethylenediamine-tetraacetic acid (EDTA) anticoagulated blood were isolated by Lympholyte-H (Cedarlane, Burlington, Canada).
p63RhoGEF protein expression
p63RhoGEF protein expression assessment was performed using Western blot analysis as previously reported. 8 Total protein extracts were obtained by cell lysis with ice-cold buffer (Tris HCl 20 mmol/l, NaCl 150 mmol/l, EDTA 5.0 mmol/l, Niaproof 1.5%, Na3VO4 1.0 mmol/l, sodium dodecyl sulfate (SDS) 0.1%, phenylmethylsulfonyl fluoride (PMSF) 0.5 mmol/l with added protease inhibitors; Complete Protease Inhibitor Cocktail; Roche Diagnostics, Mannheim, Germany).
Proteins were separated by SDS-polyacrylamide gel electrophoresis (PAGE), transferred onto nitrocellulose membranes (Hybond ECL; Amersham, Uppsala, Sweden) and blocked overnight with no-fat milk (5% in Tween-phosphate-buffered saline (PBS)). Membranes were probed with primary polyclonal antip63Rho- GEF antibody (1:1000) (ProteinTech, Chicago, IL, USA) as described by Momotani et al. 6 After incubation with the appropriate horseradish peroxidase (HRP)-conjugated secondary antibodies (Amersham Biosciences, Uppsala, Sweden), immunoreactivity was visualized with chemiluminescence using Super- Signal WestPico Chemiluminescent Substrate (Pierce, Rockford, IL, USA). Glyceraldehyde 3-phosphate dehydrogenase (GAPDH) was used as a loading control (Millipore, Temecula, CA, USA). p63RhoGEF protein expression was quantified using a densitometric semiquantitative analysis using United States National Institutes of Health (NIH) image software, and normalized to the GAPDH loading control.
Analysis of MYPT-1 phosphorylation
MYPT-1 phosphorylation was evaluated using Western blot analysis as previously reported. 7 Total protein extract was obtained by lysis of mononuclear cells with lysis buffer (Tris HCl 20 mmol/l, NaCl 150 mmol/l, EDTA 5.0 mmol/l, Niaproof 1.5%, Na3VO4 1.0 mmol/l, SDS 0.1%, PMSF 0.5 mmol/l) added with Proteases Inhibitor Cocktail (Roche Molecular Biochemicals, Mannheim, Germany) and Phosphatase Inhibitor Cocktail I (Sigma-Aldrich, St. Louis, MO, USA). Protein concentration was evaluated by bicinchoninic acid assay (BCA Protein Assay; Pierce). The proteins were separated by SDS-PAGE in Tris pH 8.3. Protein transfer on nitrocellulose membranes was performed using Hoefer TE 22 Mini Tank Transphor Unit (Amersham Pharmacia Biotech, Uppsala, Sweden) with use of the following transfer buffer: 39 mmol/l glycine, 48 mmol/l Tris base, 0.037% SDS (electrophoresis grade), and 20% methanol. The membranes were incubated overnight with primary polyclonal antiphospho-MYPT (Thr853) antibodies (Cell Signaling, Danvers, MA, USA) and anti-MYPT (Cell Signaling). HRP-conjugated secondary antibodies were used (Amersham Pharmacia, Uppsala, Sweden) and immunoreactive proteins were visualized with chemiluminescence using SuperSignal WestPico Chemiluminescent Substrate (Pierce).
MYPT-1 phosphorylation was evaluated using a densitometric semiquantitative analysis using NIH image software. The ratio between phospho-MYPT-1 and MYPT-1 was used as an index of MYPT-1 phosphorylation.
Statistical analysis
Data are expressed as mean±SD and analyzed using SPSS 20 for Mac (SPSS Italy Inc, Bologna, Italy) running on a Mac Pro (Apple, Cupertino, CA, USA). The data were evaluated using one-way within-subjects analysis of variance (ANOVA) followed by Bonferroni’s post hoc test. Chi-square analysis was used to assess the differences of p63RhoGEF protein expression level and MYPT-1 phosphorylation status between baseline and at three and six months. Values at less than 5% level (p < 0.05) were considered significant.
Results
In hypertensive patients the treatment with olmesartan normalized BP since the third month of treatment (BP ⩽ 140/90 mmHg) in all 15 patients (ANOVA: p < 0.001): 147±5.2 mmHg at baseline vs 136.8±2.00 at three months, p < 0.001 with a further reduction to 134.3±2.3 at six months p < 0.001 and p < 0.01 vs three months, for SBP and 96.6±2.6 at baseline vs 88.25±2.0 at three months, p <0.001 and 84.2±1.3 at six months, p < 0.001 and p < 0.01 vs three months for DBP (Figure 1).
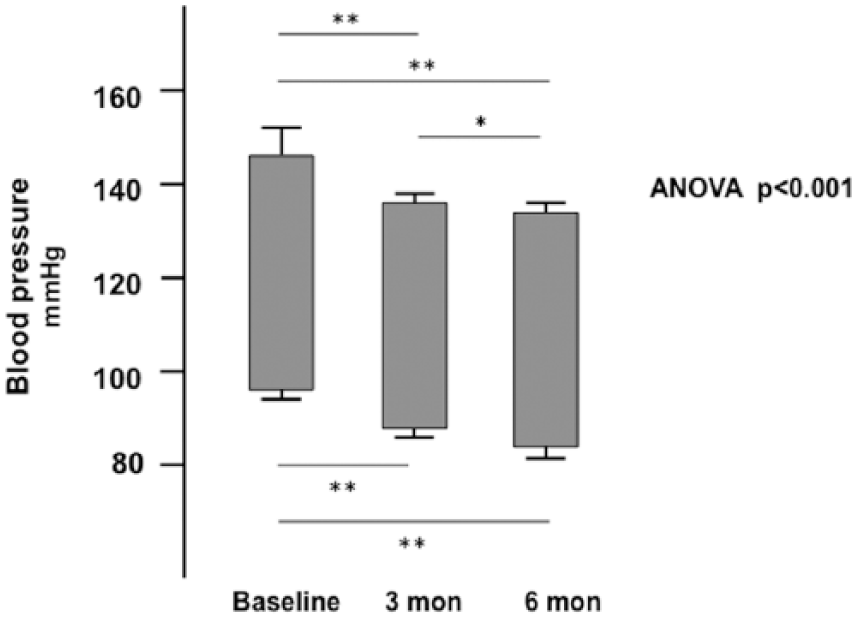
Effect of olmesartan treatment on systolic and diastolic blood pressure in the hypertensive patients enrolled in the study.
Effect of the treatment with olmesartan on p63RhoGEF protein level
Figure 2 shows that in hypertensive patients olmesartan treatment significantly reduced p63RhoGEF protein level (F = 26.32, p < 0.0001: 1.3±0.25 d.u. (baseline) vs 1.0±0.29 (three months), p < 0.0001 vs 1.0±0.22, (six months), p < 0.0001, with no difference between three and six months of treatment (p = 0.26).

Densitometric analysis of p63RhoGEF protein expression in mononuclear cells of essential hypertensive patients treated with olmesartan.
Effect of olmesartan on MYPT-1 phosphorylation status
Figure 3 shows that in hypertensive patients olmesartan treatment significantly reduced MYPT-1 phosphorylation (F = 46.61, p < 0.0001: 1.2 ±0.14 (baseline) vs 0.9±0.19 (three months), p = 0.008, vs 0.8±0.16 (six months), p = 0.001, with no difference between three and six months of treatment (p = 0.78).

Densitometric analysis of phospho-MYPT-1 to MYPT ratio in mononuclear cells of essential hypertensive patients treated with olmesartan.
Discussion
The complexity of the Ang II signaling pathways leading to vasoconstriction, hypertension, cardiovascular-renal remodeling and atherothrombogenesis has been recently expanded by the evidence of a major role played by RhoA/Rho kinase activation-triggered processes.1,9 A critical role for the transduction of Ang II signaling to directly activate the RhoA/Rho kinase pathway has been provided for p63RhoGEF as demonstrated in vitro by studies of loss of function and functional responses5,7 and supported by reports of its increased level in animal models of hypertension 20 and in hypertensive patients.8,21
One of the processes triggered by RhoA/Rho kinase activation is the Ang II-mediated increase of oxidative stress via increased expression of nicotinamide adenine dinucleotide phosphate (NADPH) oxidase 10 with subsequent endothelial dysfunction, production of proinflammatory cytokines and growth factors ultimately leading to cardiovascular and renal remodeling, target organ damages of hypertension.
Among antihypertensive drugs, olmesartan, an Ang II AT1R blocker, widely used in the treatment of hypertension, has been shown to possess antioxidant and activating NO system-related effects.11-14 In addition, using a molecular biology approach, we have reported in essential hypertensive patients that six-month olmesartan treatment reduced the protein expression of p22phox, a subunit of NADPH oxidase essential for the induction of oxidative stress, 22 reduced the phosphorylation state of extracellular signal-regulated protein kinases 1 and 2 (ERK1/2), effector of oxidative stress-mediated Ang II signaling for cardiovascular remodeling, and reduced the plasma level of oxLDL, a marker of oxidative stress. 15 Moreover, the treatment of these patients with olmesartan increased the protein expression of heme oxygenase-1, 15 a potent antioxidant and anti-inflammatory protein,16,17 and the number of cEPCs and reduced cEPCs apoptosis in addition to increase the level of calcitonin gene-related peptide, 18 a vasorelaxant that prevents cEPCs senescence, reverses Ang II-induced senescence of EPCs and reduces blood pressure in spontaneously hypertensive rats. 19 Furthermore, although indirectly, the vasoprotective, anti-inflammatory and anti-atherosclerotic effects of olmesartan shown in humans in the European Trial on Olmesartan and Pravastatin in Inflammation and Atherosclerosis (EUTOPIA), Vascular Improvement with Olmesartan medoxomil Study (VIOS), Multicenter Olmesartan Atherosclerosis Regression Evaluation (MORE) and Impact of OLmesartan on progression of coronary atherosclerosis: Evaluation by IntraVascular UltraSound (OLIVUS) clinical trials23–26 can be linked with an inhibitory effect on oxidative stress and oxidative stress signaling as a mechanism involved in the effects of olmesartan observed in these studies. Given these effects of olmesartan on oxidative stress and on the improvement of endothelial dysfunction including inflammatory processes mediated by Ang II and given that RhoA/Rho kinase activation, as mentioned above, is mainly involved in the Ang II signaling following Ang II AT1R activation including oxidative stress,1–3,9 it comes as no surprise that olmesartan may be able to downregulate the RhoA/Rho kinase pathway via AT1R blockade and reduction of oxidative stress effects. This is exactly what is reported by the results of the present study, which show in hypertensive patients on olmesartan treatment a reduced protein level of p63RhoGEF, the transducer of Ang II message to directly activate RhoA/Rho kinase pathway 5 and a reduced phosphorylation state of MYPT-1, the regulatory subunit of MLC phosphatase (MLCP), the target of Rho kinase-mediated inhibitory phosphorylation of MLCP, this latter resulting in the inhibition of the Rho kinase-induced increased activity of MLC kinase leading to reduced vascular smooth muscle contraction and proliferation. 2
Although the extrapolation of these data obtained in mononuclear cells to mechanisms that usually take place in endothelial and smooth muscle cells might be considered a limitation, it should be noted that circulating blood cells are widely used in vascular biology to study ex-vivo pathophysiological mechanisms of hypertension and remodeling. 27 In addition, the role of various inflammatory mechanisms such as mononuclear leukocyte infiltration for the development of hypertensive target organ damage has been increasingly recognized in the last few years 27 and a correlation between leukocyte (polymorphonuclear and mononuclear cells) intracellular oxidative stress and hypertension has recently been demonstrated. 28 Furthermore, leukocyte Rho kinase has been shown to increase leukocyte infiltration into the vascular wall resulting in pro-inflammatory cytokines, 29 and an increase in Rho kinase activity in peripheral leukocytes has been shown in patients with hypertension. 30 These types of cells are, therefore, useful tools to investigate processes involved in hypertension, oxidative stress, and remodeling as vascular smooth muscle or endothelial cells with the advantage of being readily accessible.
The results of this study are further strengthened by the reduced p63RhoGEF protein level and reduced phosphorylation of MYPT-1 we recently documented in a human model of endogenous antagonism of Ang II signaling via AT1R with a clinical and molecular picture opposite of hypertension such as in Bartter’s/Gitelman’s patients, 9 compared to essential hypertensive patients.8,21 As Ang II directly activates RhoA/Rho kinase via AT1R stimulation/p63RhoGEF actions, the combination of the findings in Bartter’s/Gitelman’s patients,3,8 human model opposite of hypertension including the activation of Ang II signaling via Ang II type 2 receptor (AT2R),9,31 with those obtained in essential hypertensive patients on olmesartan treatment in this study, offers additional support for the proposed role of Ang II signaling via AT2R in the cardiovascular protective effects of AT1R blockers beyond AT1R blockade. 32 Thus, the inhibition of Ang II-mediated RhoA/Rho kinase activation as shown in this study by olmesartan could well be included in the cardiovascular protective effects of this drug.
It has to be noted that still unknown factors other than Ang II and vasoactive proteins might be involved in influencing ROCK activity. This could be the case, shown in animals, of low renin/hypervolemic hypertension, in which ROCK activity is increased despite low levels of Ang II, and the case of the reduction of ROCK activity in hypertensive patients on the calcium channel blocker amlodipine treatment, 30 although in this latter case a possible inhibitory effect on calcium entry in the cell, via an L-type voltage-dependent calcium channel, available for calcium sensitization by ROCK, might play a role. 33
In conclusion, this study demonstrates in hypertensive patients olmesartan’s inhibitory effect on the RhoA/Rho kinase pathway likely via the reduction of oxidative stress signaling. This effect joined with the available evidence coming from the results of clinical trials in humans, further provides a mechanistic rationale for olmesartan’s antioxidant and anti-inflammatory potential translation, in the long term, toward anti-atherosclerotic and anti-remodeling effects.
Footnotes
Acknowledgements
The authors are grateful to the nonprofit Foundation for Advanced Research in Hypertension and Cardiovascular Diseases (F.O.R.I.C.A.), Padova, Italy, for its support.
The results of this study were presented as a poster presentation at the 18th Annual Meeting of the European Council for Cardiovascular Research (ECCR), October 24–26, 2014, Poiano, Lake Garda, Italy.
Conflict of interest
None declared.
Funding
This study has been supported in part by an unrestricted research grant from Daiichi-Sankyo Italy to LAC.