
Abstract
Introduction:
Angiotensin-converting enzyme 2 (ACE2) is a new member of the renin–angiotensin system (RAS) and it has been proposed that ACE2 is a potential therapeutic target for the control of cardiovascular disease. The effect of losartan on the ACE2 activity in atherosclerosis was studied.
Methods:
Atherosclerosis was induced in New Zealand white rabbits by high-cholesterol diet for 3 months. An Angiotensin II (Ang II) receptor blocker (losartan, 25 mg/kg/d) was given for 3 months. ACE2 activity was measured by fluorescence assay and the extent of atherosclerosis was evaluated by H&E and Oil Red O staining. In addition, the effect of losartan on ACE2 activity in smooth muscle cells (SMCs) in vitro was also evaluated.
Results:
Losartan increased ACE2 activity in atherosclerosis in vivo and SMCs in vitro. Losartan inhibited atherosclerotic evolution. Addition of losartan blocked Ang II-induced down-regulation of ACE2 activity, and blockade of extracellular signal-regulated kinase (ERK1/2) with PD98059 prevented Ang II-induced down-regulation of ACE2 activity.
Conclusions:
The results showed that ACE2 activity was regulated in atherosclerotic plaque by losartan, which may play an important role in treatment of atherosclerosis. The mechanism involves Ang II–AT1R-mediated mitogen-activated protein kinases, MAPKs (MAPKs) signaling pathway.
Introduction
Several lines of evidence have indicated that the renin–angiotensin system (RAS) plays an important role in the progression of atherosclerosis.1,2 Angiotensin-converting enzyme (ACE) and Angiotensin II (Ang II) have been found in stable and unstable ruptured human coronary plaques. The localization of Ang II with positive macrophages has also been observed in the shoulder region of coronary atherosclerotic plaques and in close proximity to presumed ruptured sites of human coronary arteries with acute myocardial infarction. Thus this indicates a relationship between the RAS and atherosclerosis. Angiotensin-converting enzyme 2 (ACE2) is the newest member of the RAS and shares approximately 40% similarity with the somatic form of ACE. ACE2 catalyzes the formation of angiotensin 1–7 (Ang-(1–7)) from Ang II, and it has been proposed that ACE2 is a potential therapeutic target for the control of cardiovascular disease including atherosclerosis and coronary heart disease.3,4 Previous studies have shown that ACE and Ang II type 1 receptor (AT1R) expression have been found in atherosclerotic tissues. Furthermore, the inhibition of either ACE activity or AT1R activation by selective pharmacotherapy greatly reduced the formation of atherosclerosis. Another study found that blockade of the AT1 receptor resulted in a significant increase in ACE2 expression in the myocardium after myocardial infarction. 5 Our previous study demonstrated that the AT1R antagonist losartan not only enhanced ACE2 activity in myocardium in diabetic cardiomyopathy, but also improved cardiac function and inhibited myocardial fibrosis. This result suggests that losartan may improve left ventricular remodeling by increasing ACE2 activity, apart from its basic function of hindering Ang II binding to AT1R. 6 However, little is known about the change in ACE2 activity in atherosclerotic plaques after using AT1R antagonist.
In this study, we hypothesized that losartan can increase ACE2 activity as a potential mechanism in anti-atherosclerosis. In order to test the hypothesis, this study was carried out in two parts. First, we observed the effect of the AT1R antagonist losartan on ACE2 activity in the aortas of a rabbit model of hypercholesterolemia diet-induced atherosclerosis. Second, we explored the mechanism and signaling pathway of influencing ACE2 activity through losartan in vascular smooth muscle cells (SMCs) in vitro.
Methods
Animal model
For this study, 27 male New Zealand white rabbits (weight approximately 1.5 kg) were housed individually and assigned randomly to three regimens for 12 weeks: (1) Control group (rabbits fed with standard chow, n=9); (2) high-cholesterol diet group (HC diet group, rabbits fed with cholesterol-enriched chow containing 1% cholesterol and 5% peanut oil, n=9); and (3) high-cholesterol diet + losartan group (losartan group, rabbits fed with cholesterol-enriched chow and losartan, 25 mg/kg/day in drinking water, n=9). The consumption of water was measured every other day to confirm the appropriate administration of the drug. The rabbits were fed for 3 months. The animal experimental protocol complied with the Animal Management Rules of the Chinese Ministry of Health (document No 55, 2001) and was approved by Animal Care Committee of Shandong University.
Histology
Consecutive segments of the abdominal aorta were embedded in paraffin or OCT, sectioned, mounted on glass slides, and conventionally stained with H&E and Oil Red O. Serial sections with the maximal amount of lesions were chosen for quantification by a single observer blinded to the experimental protocol.
Immunohistochemistry
At the end of the experiment, animals were deeply anesthetized with a mixture of ketamine hydrochloride (40 mg/kg IM) and pentobarbital sodium (50 mg/kg IV). The aorta was removed and immersed in 10% phosphate-buffered formalin.
Paraffin-embedded arteries were cross-sectioned into pieces 4 µm thick at 5-mm intervals from the proximal to the distal end, dewaxed, and rehydrated. Macrophages were identified with a monoclonal antibody for rabbit macrophages (1:200, RAM-11, DAKO Corp, USA). SMCs were identified with a mouse anti-human a-SM actin (1:100, DAKO Corp, USA). Monocyte chemotactic protein-1 (MCP-1) and ERK1/2 were detected with a polyclonal goat anti-human antibody (1:100, Abcam, USA). ACE2 was detected with a polyclonal goat antibody (1:100, Abcam, USA). Ang-(1–7) was identified with a polyclonal antibody against Ang-(1–7) antibody (1:100, Phoenix Pharmaceuticals, Inc, Belmont, CA, USA). After incubation with biotinylated secondary antibody followed by avidin-biotin amplification for 30 minutes, an avidin-biotin-peroxidase complex was added to the section and incubated for 30 minutes. The slides were then incubated with 3, 39-diaminobenzidine (DAB) and counterstained with hematoxylin. In each experiment, negative controls without the primary antibody or with an unrelated antibody were included to check for nonspecific staining. The arterial cross-sections stained with the antibodies were digitized and analyzed by a MSF-300G Scanner (Microtek Lab, Nikon, Japan). Quantification of ACE2 and Ang1–7 expression involved the use of an automated image analysis system (Image-Pro Plus 5.0, Media Cybernetics), and the positive staining area was measured and expressed as a mean percentage of the lesion area in at least 10 high-power fields (×400 magnification).
Determination of ACE2 activity
ACE2 activity was measured by an enzymatic assay. ACE2 fluorescence assay was based on the use of the fluorogenic peptide substrate V (FPS V, 7-Mca-RPPGFSAFK(Dnp)-OH, R&D Systems). ACE2 cleaves the C-terminal dinitrophenyl moiety and results in increased fluorescence at excitation and emission spectra of 320 and 405 nm, respectively.7–9 Cells or atherosclerotic plaque samples were homogenized and the protein concentration was measured; tissue samples were diluted in a buffer. To each well, the diluted plaque tissue with buffer was added, along with 10 μl of buffer (with the respective inhibitor), and the reaction was initiated by the addition of the substrate. The total volume for each well was 100 μl. All reactions were performed at room temperature for 18–24 h. The plates were read using a fluorescence reader (Turner Biosystems, USA) and ACE2 activity was measured. Specific ACE2 activity was measured by A–B, where A is the fluorescence in the presence of the ACE inhibitor, captopril, and B is the fluorescence in the presence of the ACE inhibitor and ACE2 inhibitor, DX600. All reactions were performed at ambient temperature in microtiter plates.ACE2 activity was expressed as RFU/μg/hour.
Level of Ang-(1–7) by ELISA
The concentration of Ang-(1–7) in the atherosclerotic plaque was measured by a commercial enzyme-linked immunosorbent assay (ELISA) kit (Bachem, USA). In brief, the atherosclerotic plaque was mechanically homogenized on ice. Homogenized samples were centrifuged at 10,000 rpm for 10 min at 4°C. Each supernatant was stored at −80°C, and the level of Ang-(1–7) was measured by ELISA.
Western blot
ACE2 protein expression from membrane protein preparations was detected in SMCs and atherosclerotic plaque. ERK1/2 protein expression from total protein preparations was detected in atherosclerotic plaque. Equal amounts of protein per well (50 µg protein) were transferred onto pure nitrocellulose membranes. Membranes were incubated with primary antibodies (1:200, Santa Cruz Biotechnology, USA) overnight at 4°C. The next day, membranes were exposed to the secondary antibody conjugated with horseradish peroxidase (1:2000), and films were incubated with enhanced chemiluminescence substrate. The densities of the bands were analyzed using a MSF-300G Scanner (Microtek Lab, Nikon, Japan).
SMC cell culture
SMCs were cultured from human umbilical artery. Briefly, adhering connective tissue was removed by blunt dissection from the umbilical artery. The umbilical arteries were opened longitudinally, minced into 1-mm pieces, incubated for about 1.5–2 h, and rinsed twice with PBS to remove the cells, which were seeded in plastic culture flasks. Cells were harvested for passage at 2-week intervals and used between the 2nd and 3rd passages. The cells exhibited the typical ‘hill and valley’ growth morphology of SMC and reacted with the monoclonal antibody which selectively recognizes muscle forms of α-actin.
SMCs were plated at a density of 2.5×104 cells per well in 24-well tissue culture dishes and allowed to grow to confluence in DMEM/F12 medium containing 10% FCS under standard culture conditions. For all experiments, the cells were made quiescent by incubation in serum-free DMEM/F12 for 24 h before stimulation with Ang II (10−6 mol/L) for 24 h.
To inhibit the binding of Ang II to its type I receptor, losartan (10 µmol/l) was used. To inhibit Ang II-induced ERK1/2 kinase, inhibitor to ERK1/2 kinase (PD98059, 20 µmol/l, Cell Signaling Technology, Danvers, MA) was used.
SMCs were incubated with Ang II for 24 h before they were divided into the following groups that received losartan (10 µmol/l), Ang II (10−6mol/l), Ang II +ERK1/2 inhibitor, and no treatment (control group), respectively.
Statistical analysis
Data analysis involved use of SPSS 11.5. One-way analysis of variance (ANOVA) was used to test the difference of means among three groups for continuous variables. A p-value <0.05 was considered statistically significant.
Results
Location of ACE2 and Ang-(1–7) protein expression in rabbit atherosclerotic lesions
To determine ACE2 expression and cell types in atherosclerotic plaques, adjacent sections were used for immunohistochemical staining of antibodies against RAM-11, a-SM actin and ACE2. The results showed positive ACE2 protein expression in macrophages, VSMCs (Figure 1 (a1, a2) and (b1, b2)) and aortic endothelia (Figure 1(c, d)). Furthermore, Ang-(1–7), the enzymatic product of ACE2, was found to co-localize with ACE2 in atherosclerotic lesions (Figure 1(e1, e2) and (f1, f2).
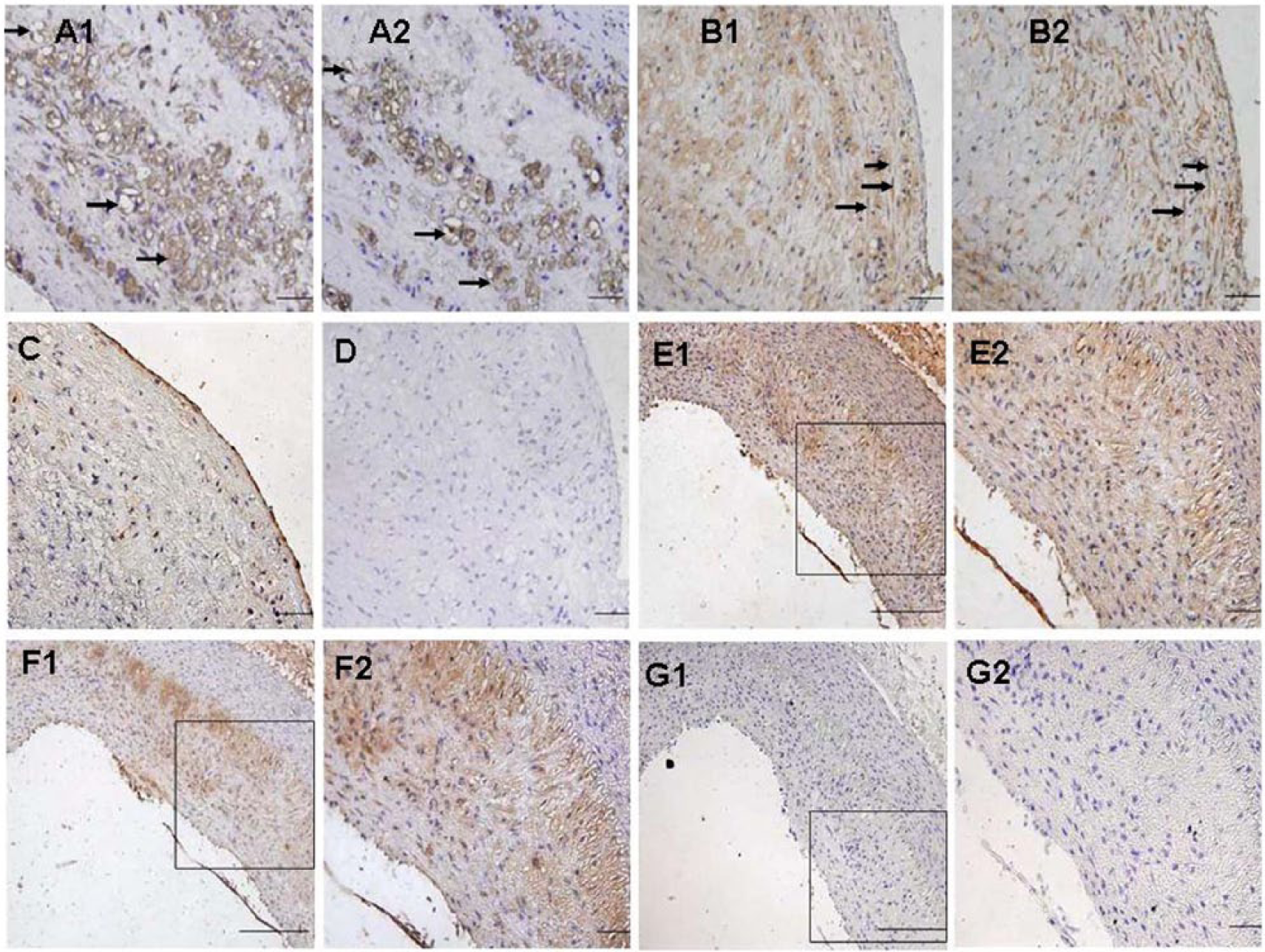
Effect of losartan on atherosclerosis
There was no intimal and medial thickening in control group. The intimal thickening and ratio of intima / media were significantly higher in the HC diet group than those in the control group (Figure 2(a, b) p<0.01). However, the intimal thickening and ratio of intima / media were significantly decreased in losartan group compared with the HC diet group (Figure 2(a, b), p<0.05). As we can see, sections of control group did not show any lipid staining. The lipid area was significantly higher in the HC diet group than in losartan group (Figure 2(c, d) p<0.05).
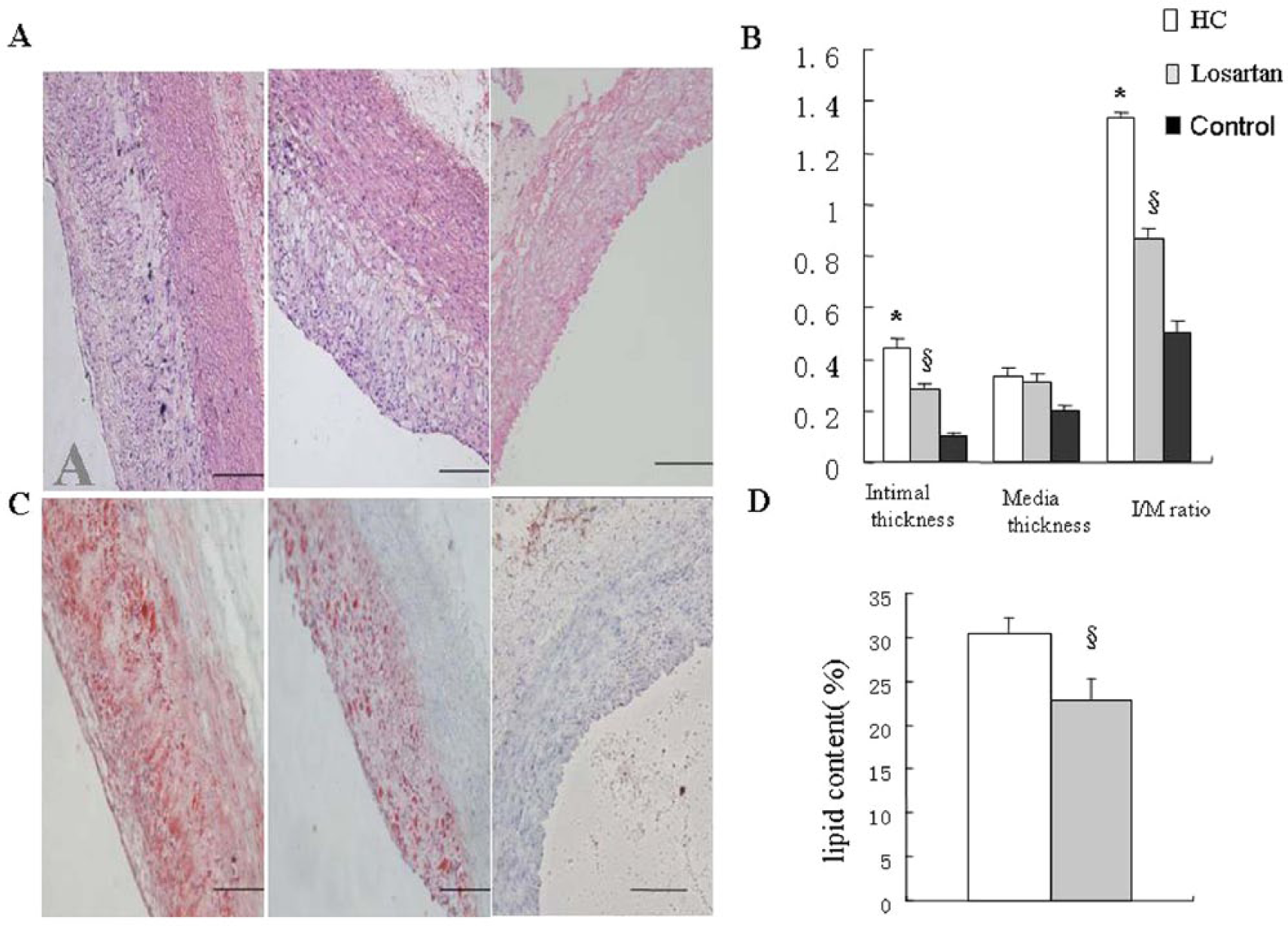
Losartan inhibited atherosclerotic plaque evolution.
The level of ACE2 activity, ACE2 protein and Ang-(1–7) protein expression in atherosclerotic plaques by losartan
ACE2 activity in the losartan group (2.66±0.19 U/μg protein/h) and the HC diet group (1.30±0.18 U/μg protein/h) was markedly higher than that in the control group (0.29±0.07 U/μg protein/h, p<0.05). Furthermore, ACE2 activity was further increased in the losartan group compared with the HC diet group (p<0.05). Similarly, the level of ACE2 protein in aortic plaque by immunohistochemistry and Western blot was significantly higher in the losartan group and HC diet group than in control group (Figure 3(a, b, d, e), p<0.01). Furthermore, the level of ACE2 protein was further increased in losartan group compared with the HC diet group (p<0.01). The level of Ang-(1–7) protein in aortic plaque by immunohistochemistry and ELISA was also higher in the losartan group and HC diet group than in control group (Figure 3(a, c, f), p<0.01). In contrast, the level of Ang-(1–7) protein was statistically higher in the losartan group than in HC diet group (Figure 3(a, c, f), p<0.01).
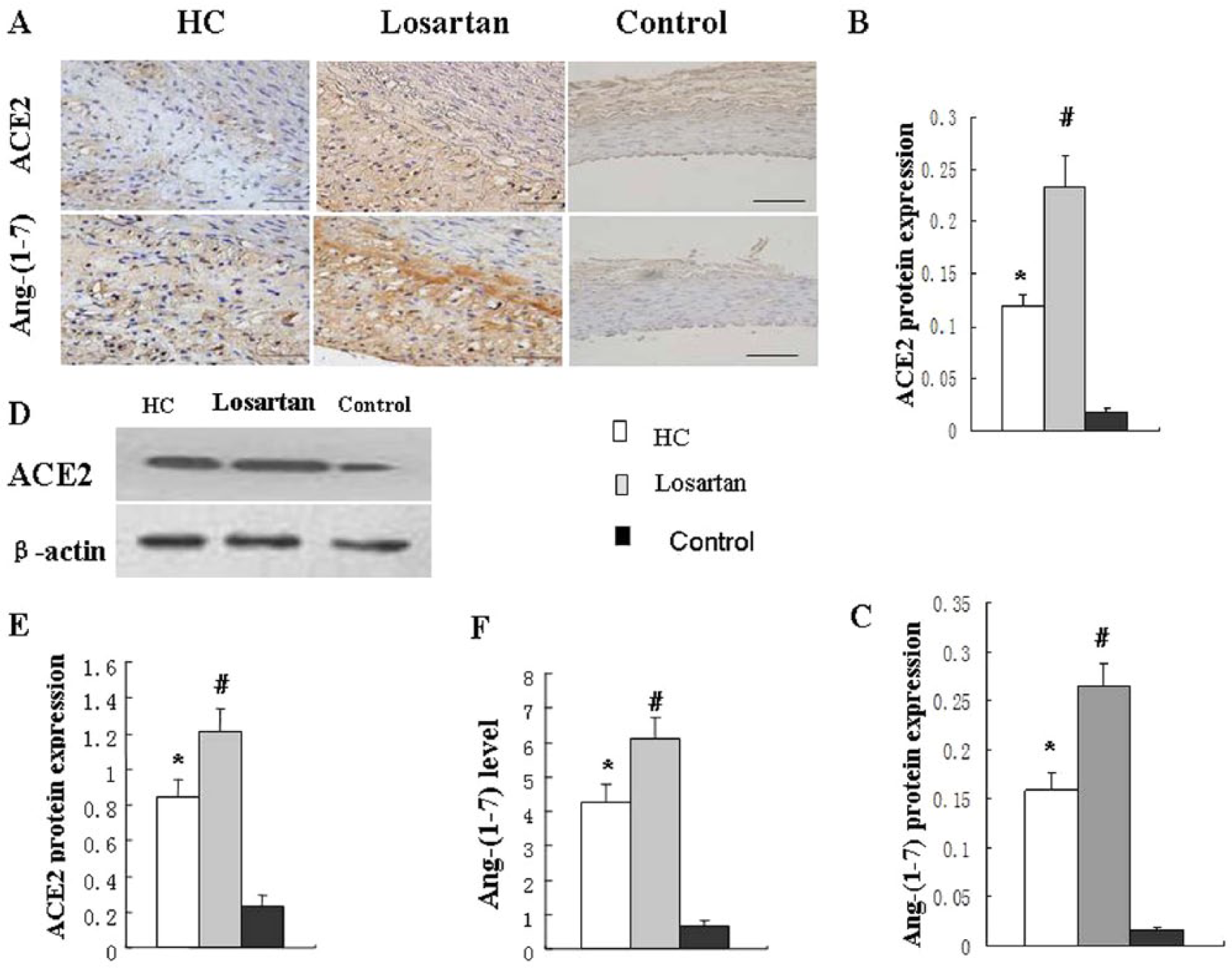
Losartan increased ACE2 and Ang-(1–7) protein expression in atherosclerotic plaque.
Losartan prevents Ang II down-regulation of levels of ACE2 protein and ACE2 activity in vitro
Cultured VSMCs were stimulated with Ang II to detect whether Ang II regulates ACE2 protein expression and ACE2 activity. The results showed that Ang II significantly down-regulated ACE2 protein expression (Figure 4(a, b)), and activity (Figure 4(c)). Losartan (10 µmol/l) effectively blocked Ang II-modulated reduction of ACE2 protein (Figure 4(a, b)) and activity (Figure 4(c)).

ACE2 protein expression and activity in SMCs.
Signaling mechanism of losartan preventing Ang II down-regulation of ACE2 activity
The ERK1/2 signaling pathways were explored in vivo and in vitro. ERK1/2 protein expression was evaluated by immunohistochemistry (Figure 5(a)) and Western blot (Figure 5(b)). The results showed that ERK1/2 protein was weakly positive in the control group. On the contrary, the ERK1/2 protein was strongly expressed in HC diet group. Administration of losartan in the losartan group resulted in weak expression of the ERK1/2 protein. ERK1/2 phosphorylation in atherosclerotic lesions was significantly lower in the losartan group than in the HC diet group (Figure 5(c), p<0.01).

ERK1/2 protein expression.
Cultured VSMCs were incubated with Ang II for 24 h in the presence or absence of PD98059 (20 μmol/l), an inhibitor of ERK1/2 MAP kinase, to investigate the cellular signaling pathways involved in the reduction of ACE2 activity induced by Ang II. PD98059 (20 µmol/l) blocked the Ang II-mediated decrease in ACE2 activity (Figure 4(d), p<0.01). Treatment of the VSMCs with PD98059 alone had no effect. Losartan blocked Ang II-induced decrease of ACE2 protein and activity. These results suggested that down-regulation of ACE2 induced by Ang II is mediated by a MAP kinase signaling pathway. Losartan prevented Ang II-induced decrease of ACE2 protein and activity.
Discussion
This study demonstrated that ACE2 protein was expressed in macrophages, endothelial cells and SMCs in atherosclerotic plaque in a rabbit model; and Ang-(1–7) was also expressed in plaque. Losartan, an AT1 receptor blockade, not only increases ACE2 activity, but also increases Ang-(1–7) protein expression. Furthermore, our in vitro study showed that losartan blocked Ang II-induced decrease of ACE2 activity and blockade of ERK1/2 with PD98059 prevented Ang II-induced down-regulation of ACE2 activity. The results suggested that losartan increases ACE2 activity by preventing the Ang II–AT1R-mediated ERK/ MAP kinase signaling pathway.
A series of evidence demonstrated that the RAS plays an important role in the pathogenesis of cardiovascular disease, especially atherosclerosis and coronary heart disease.10,11 Previous research reported that Ang II was localized at the shoulder region of coronary atherosclerotic plaques and in close proximity to the rupture site of coronary arteries in unstable angina and acute myocardial infarction. Some studies have reported that Ang II stimulates the production of MCP-1 and adhesion molecules such as vascular cell adhesion molecule 1 (VCAM-1), intracellular adhesion molecule 1, and (ICAM-1); Ang II also induces the production of matrix metalloproteinase 3 (MMP-3) and matrix metalloproteinase 9 (MMP-9) in macrophage and SMCs; thus Ang II takes part in the pathogenesis of atherosclerosis. 12
ACE2 is a newly discovered member of the RAS. Recent studies have shown that it is a potential therapeutic target for the treatment of atherosclerosis, coronary heart disease and diabetic cardiomyopathy owing to its key role in the formation of vasoprotective peptides, Ang-(1–7), from Ang II.3,13 Our study and other studies showed that ACE2 and Ang-(1–7) inhibit early atherosclerosis and increase stability of atherosclerotic plaques. Our study showed that ACE2 overexpression attenuated the progression of lesions and stabilized atherosclerotic plaques in rabbits, which were formed by endothelial injury and an atherogenic diet. 14 Sahara et al. reported that ACE2 gene knock-out in mouse increased the severity of atherosclerosis and produced an inflammatory response in plaque, suggesting that ACE2 is an important role in anti-atherosclerosis. 15 Thatcher et al. found that ACE2 deficiency increased atherosclerotic area and inflammatory response through regulation of the ratio of Ang II/Ang-(1–7) peptides. 16 Langeveld et al. reported that Ang-(1–7) attenuated new intima development of atherosclerosis and prohibited atherosclerotic evolution. 17 These studies suggested that the higher level of ACE2 and Ang-(1–7) in atherosclerosis have a beneficially therapeutic effect in the pathogenesis of atherosclerosis. Our results showed that administration of losartan increased ACE2 activity and protein production in the plaque. We also found that losartan inhibited evolution of atherosclerotic plaque, which is associated with the increased ACE2 protein and ACE2 activity in the plaque. This result suggested that ACE2 may be responsible for the beneficial treatment of atherosclerosis and may be an important role in anti-atherosclerotic plaque formation using losartan. Ferrario et al. found that losartan increased cardiac ACE2 mRNA and cardiac ACE2 activity and levels of both Ang II and Ang-(1–7) in Lewis rats. 18 Xia et al showed that Ang II type 1 receptor mediated ACE2 inhibition and losartan increased central ACE2 activity in chronically hypertensive mice. 19 Ishiyama et al. investigated ACE 2 expression in the model of myocardial infarction in Lewis normotensive rats by using losartan, and found that losartan not only increased cardiac ACE2 mRNA, but also induced partial reversal of left ventricular remodeling and left ventricular hypertrophy. 5 Our previous study showed that losartan increased ACE2 protein and activity in diabetic cardiomyopathy in rats. In the meanwhile, losartan also improved left ventricular remodeling, and this result suggested that increased ACE2 activity by losartan may be an important factor in the influence of cardiac function and left ventricular remodeling in diabetic cardiomyopathy. With the combination of this study and our data, we think that ACE2 may contribute a beneficial effect in the treatment of the coronary disease by using losartan, which may be a new factor in using losartan.
Ang-(1–7) is an important effective treatment factor in the pathogenesis of atherosclerosis and heart disease. Sampaio et al. found that Ang-(1–7) regulated nitric oxide production through PI3K/Akt-dependent pathways in endothelial cells in vitro, increased endothelial-dependent vasodilator function, and protected endothelial function. 20 Strawn et al. found that Ang-(1–7) inhibited SMC proliferation and migration and prohibited neointimal formation in balloon-injured carotid arteries in rat. 21 Our data showed that losartan increased Ang-(1–7) protein expression. This change was in parallel with augmented ACE2 activity and ACE2 protein. The results suggested that increased ACE2 activity induced more conversion from Ang II to Ang-(1–7).
Our in vitro study showed that addition of losartan blocked Ang II-induced down-regulation of ACE2 protein and activity. Furthermore, our study found that blockade of ERK1/2 with PD98059 prevented Ang II-induced down-regulation of ACE2 activity, and the result further demonstrated that the Ang II–AT1R-mediated ERK/ MAP kinase signaling pathway may be an important mechanism by which Ang II down-regulates ACE2 expression. Losartan prevented the AT1R-mediated ERK/P38 MAP kinase signaling pathway and further prevented Ang II down-regulation of ACE2 expression. A study by Gallagher et al. also found that Ang II down-regulated ACE2 mRNA; losartan and Ang-(1–7) blocked the effect of Ang II-mediated reduction of ACE2 mRNA. 22 Our previous study showed that AngII induced ERK-p38 activation in vitro and ACE2 overexpression inhibited atherosclerosis and down-regulated ERK-p38 signaling pathways. 14 Furthermore, our study found that ERK1/2 phosphorylation in atherosclerotic lesions was significantly higher in the HC diet group than in losartan group; this indicated that administration of losartan decreased ERK1/2 protein expression. Combining our study and the study by Gallagher et al., we speculate that MAPKs signaling pathways may play an important role in the regulation of ACE2 by AngII. Losartan increased ACE2 expression and up-regulated ACE2 activity by prevention of AT1R-mediated MAPKs signaling pathway.
The factors and mechanisms for the change in ACE2 activity in atherosclerotic plaque are not very clear and are rather complicated, but a recent study showed that ACE2 activity undergoes a dynamic change during atherosclerotic plaque evolution. The RAS system is highly activated in the pathogenesis of atherosclerosis. ACE and AngII were expressed in endothelial cells, SMCs and macrophages. Similarly, our previous study and other study demonstrated that ACE2 protein was also expressed in endothelial cells, SMCs and macrophages in atherosclerotic plaque.14,23 Sluimer et al. found that ACE2 protein was present in atherosclerotic carotid arteries and that ACE and ACE2 protein were expressed in total vessel wall during all stages of atherosclerosis. Furthermore, they also found that ACE2 activity was lower in stable atherosclerotic plaque compared with vulnerable plaque, suggesting that ACE2 activity was regulated in atherosclerosis in different period and progression of atherosclerotic plaque. 23 Our study showed that AngII was involved in the down-regulation of expression/activation of ACE2 in the in vitro study. Other research has shown that hyperglycemia was positively correlated with ACE2 activity. 24 From the above study, it is speculated that ACE2 activity and protein levels may vary during atherosclerotic plaque evolution. Endogenous factors, including plaque type, hyperlipidemic and hyperglycemic environment, ACE and Ang II activation, may take part in the regulation and influence of ACE2 expression/activation. Increased ACE2 activation was considered a protective and compensatory mechanism in the counterbalance of ACE activity, and may play an important role in treatment of cardiovascular diseases. 25
In conclusion, this study showed that ACE2 activity was regulated in atherosclerotic plaque by losartan, which may play an important role in the treatment of atherosclerosis. The mechanism acts on the MAPK signaling pathway.
Footnotes
Acknowledgements
We thank Zhaoli Zhou, Hong Jiang and Xuping Wang for their technical assistance.
Conflicts of interest
None declared.
Funding
This study was supported by the National 973 Basic Research Program of China (No. 2013CB530700), the National Natural Science Foundation of China (No. 81170207) and the Program of State Chinese Medicine Administration Bureau (No. JDZX2012113).