
Abstract
Introduction:
The angiotensin II (Ang II) type 1 receptor exerts pro-atherogenic action by augmenting oxidative stress, whereas the Ang II type 2 receptor (AT2)-mediated effect on atherosclerosis remains controversial.
Materials and methods:
AT2 transgenic (AT2-Tg) mice, which overexpress AT2 in their vascular smooth muscle cells, were crossed with apoE-deficient (apoE-/-) mice to generate AT2 transgenic apoE-/- mice (AT2-Tg/apoE-/-).
Results:
A subpressor dose of Ang II infusion exaggerated atherosclerosis development in apoE-/- mice, which was markedly suppressed in AT2-Tg/apoE-/- mice. Inhibitors of nitric oxide (NO) synthase (L-NAME) or bradykinin type 2 receptor completely abolished AT2-mediated anti-atherogenic actions. The vascular cell adhesion molecule-1 expression levels and degree of monocyte/macrophage accumulation in the intima were also considerably reduced in AT2-Tg/apoE-/- mice; these phenomena were completely reversed by L-NAME treatment. Ang II infusion significantly enhanced the accumulation of dihydroethidium-positive mononuclear cells in the intima and mRNA expression levels of Nox2, a phagocytic cell-type NADPH oxidase subunit in apoE-/- mice, which was completely inhibited in AT2-Tg/apoE-/- mice.
Conclusions:
Vascular AT2 stimulation exerts anti-atherogenic actions in an endothelial kinin/NO-dependent manner, and its anti-oxidative effect is likely to be exerted by inhibiting the accumulation of superoxide-producing mononuclear leukocytes.
Introduction
Angiotensin II (Ang II) has been shown to promote atherosclerosis1,2 by activating various signaling pathways and augmenting oxidative stress via type 1 receptor (AT1) activation,3,4 which leads to increased expressions of the redox-sensitive genes involved in atherogenesis.5,6 In contrast, the effect of Ang II type 2 receptor (AT2) on atherogenesis remains controversial. Daugherty et al. demonstrated that AT2 antagonist treatment exaggerated Ang II-induced atherosclerosis in apoE-/- mice 7 ; however, studies using AT2-deficient mice showed conflicting results. Daugherty et al. showed that AT2 deficiency did not affect atherosclerosis in low density lipoprotein (LDL) receptor-deficient mice, 8 whereas Iwai et al. reported that the atherosclerotic lesion area was significantly enhanced in AT2/apoE double-deficient mice through the exacerbation of oxidative stress. 9 These results raise concerns about the AT2-medited anti-atherogenic effect of long-term treatment with Ang II receptor blockers (ARB) or compound 21 (C21), a recently developed orally active non-peptide and selective AT2 agonist. 10
The expression of AT2 is known to be restricted in the adult cardiovascular system, with lower expression levels relative to the AT1 receptor, while it is up-regulated in various pathological conditions including post-myocardial infarction, heart failure, and arterial injury.11–13 It was recently shown that the AT2 is expressed in the muscular medial layer of the aorta and in endothelial cells of resistant arteries, 14 and it is also up-regulated in the aortas of apoE-/- mice on an atherogenic diet.9,15 We previously generated AT2 transgenic (AT2-Tg) mice featuring targeted AT2 overexpression in the vascular smooth muscle cells (VSMCs) and demonstrated that AT2 in VSMCs promotes intracellular acidosis by blocking the amiloride-sensitive Na+/H+ exchanger, leading to kininogenase activation followed by bradykinin release from VSMCs, resulting in endothelial nitric oxide (NO)-dependent vasodilation and an anti-pressor response to Ang II. 16 These findings lead to the hypothesis that AT2 in the aortic medial layer exerts its anti-atherogenic action through endothelial bradykinin/NO system activation.
In this study, we crossed AT2-Tg mice with apoE-/- mice to generate vascular AT2-overexpressing apoE-/- (AT2-Tg/apoE-/-) mice in an effort to elucidate the involvement of the kinin/NO system in the observed AT2-mediated anti-atherogenic action. We found that vascular AT2 activation exerts its anti-atherogenic action in an endothelial kinin/NO-dependent manner accompanied by decreased oxidative stress through decreased accumulation of superoxide-producing mononuclear leukocytes. Our findings support the notion that direct AT2 activation could be a potential therapeutic option for preventing the development of atherosclerotic cardiovascular diseases.
Materials and methods
Animal preparation
AT2-Tg mice (C57BL/6) overexpressing the AT2 under control of the α-smooth muscle actin promoter were generated as described previously. 16 Homozygous AT2-Tg mice were crossed with homozygous apoE-/- mice (C57BL/6) to yield double heterozygous mice. These heterozygous littermates were bred with apoE-/- mice to establish AT2-Tg/apoE-/- mice (backcrossed>10 times). Male apoE-/- mice and AT2-Tg/apoE-/- mice were weaned at 5 weeks of age onto a high-cholesterol diet (21% fat, 0.125% cholesterol; Oriental Yeast Co., Tokyo, Japan) that was maintained for 8 weeks. Starting at the age of 8 weeks, a subpressor dose of Ang II (100 ng/kg/min) or phosphate-buffered saline (PBS) was administered for 4 weeks using an osmotic minipump. Before the implantation of osmotic minipumps, mice were anesthetized with a single intraperitoneal injection of ketamine (50 mg/kg) and xylazine (8 mg/kg). Sufficient anesthesia depth was confirmed by the lack of a tail pinch response. Additional groups of 8-week-old apoE-/- and AT2-Tg/apoE-/- mice were treated with either L-NG-nitroarginine methyl ester (L-NAME; 1 mg/ml in drinking water) or the bradykinin receptor inhibitor icatibant (70 mg/kg/day, osmotic minipump) in parallel with the Ang II infusion.
Before the aortic tissues were isolated, the mice were sacrificed by transcardiac perfusion under terminal anesthesia by intraperitoneal injection of pentobarbital (200 mg/kg). All investigations conformed to the Guidelines for Animal Experiments of the Kyoto Prefectural University of Medicine. The experiments were also approved by a local university ethics review board.
Membrane preparation and receptor assay
Membrane fractions were prepared from pooled aortic samples and incubated with different concentrations (0.05–1 nM) of [125I]Sar1, Ile8-Ang II, or [125I]CGP42112A for 120 min at 20°C for the saturation experiment as described previously. 17 Specific binding of [125I]Sar1, Ile8-Ang II, and [125I]CGP42112A was determined from the difference between counts in the absence of 10 µmol/l AT1 antagonist (losartan) and the presence of 1 µmol/l AT2 antagonist (CGP42112A). Kd and Bmax values were estimated by Rosenthal analysis of the saturation data, while the AT1 and AT2 densities were calculated from the Bmax values.
Hemodynamic analysis
Mean blood pressure and heart rate were measured under conscious and unrestrained conditions using a programmable sphygmomanometer (BP-98A; Softron, Tokyo, Japan).
Plasma lipid analysis
Total cholesterol and triglyceride concentrations were determined using an automated chemistry analyzer (Wako Chemicals Co., Tokyo, Japan). LDL-cholesterol levels were quantified by an enzymatic reaction using a commercially available kit (Daiichi Pure Chemicals Co., Tokyo, Japan).
Quantitative measurement of atherosclerotic lesions
The mice were euthanized at 12 weeks of age and the atherosclerotic lesions were analyzed as described previously. 18 Image analysis was performed on Oil Red O-stained aortas using Scion Imaging software. The aortic lesion area in each animal was measured as the percentage of lesion area per total aortic area.
Immunohistochemistry
For immunohistochemical analysis and superoxide product evaluation, Ang II was administered for 2 weeks to the apoE-/- and AT2-Tg/apoE-/- mice from the age of 8 weeks. The aortic arch was rapidly removed after PBS perfusion, embedded in optimal cutting temperature compound, and quick-frozen in liquid nitrogen. The atherosclerotic lesions in the aortic arch region were examined at five locations at 100 µm intervals, and nine serial 6 µm-thick sections were prepared from each location, stained immunohistochemically with fluorescein isothiocyanate (FITC)- or tetramethyl rhodamine isothiocyanate (TRITC)-conjugated antibodies against vascular cell adhesion molecule-1 (VCAM-1; clone 429, Pharmingen, San Diego, CA) and monocyte/macrophage antibody-2 (MOMA-2; Serotec, Oxford, UK), and then observed using a confocal microscope (FLUOVIEW FV300; Olympus, Tokyo, Japan). As a negative control, non-immune immunoglobulin and FITC- or TRITC-conjugated secondary antibodies were used. Positive VCAM-1 and MOMA-2 staining was evaluated using image-analysis software. The percentage coverage of VCAM-1 in the total aortic area and the MOMA-2 staining area were assessed in five sections from six animals in each group.
In situ detection of superoxide anions
Fresh-frozen cross-sections of the ascending aorta (30 µm each) were incubated with dihydroethidium (DHE) (1 µM) in PBS for 30 min at 37°C in a humidified chamber protected from light. 19 To detect the ethidium, samples were examined using a fluorescence microscope equipped with a computer-based imaging system. The fluorescence intensity was pixilated and quantified using Scion Image software.
Real-time polymerase chain reaction
The thoracic aortas of the mice were dissected and the total RNA was isolated. Real-time polymerase chain reaction (RT-PCR) was performed using a LightCycler ST300 (Roche Applied Science, Indianapolis, IN) with SYBR Green PCR Master Mix and RT-PCR (Applied Biosystems, Foster City, CA) as described previously. 20 The dissociation curves were monitored to check for the aberrant primer dimers formation. PCR-amplified products were electrophoresed on 2% agarose gels to confirm the presence of a single band. Data are expressed as levels relative to apoE-/- mice with PBS treatment.
Nicotinamide adenine dinucleotide phosphate oxidase activity
Protein samples were extracted from aortas after 1 week of Ang II infusion, and incubated for 30 min in Krebs-HEPES buffer at 37°C. The rings were transferred to scintillation vials containing 100 µmol/l L-012 in Krebs-HEPES buffer and incubated for 5 min at 37°C in the dark. After incubation, chemiluminescence was measured using a luminometer (Lumat LB 9507; Berthold Technologies Ltd) over a period of 10 min at 1 min intervals. After measurement, 100 µmol/l nicotinamide adenine dinucleotide phosphate (NADPH) (Sigma Chemical Co, St Louis, MO) was added and the rings were further incubated at 37°C for 60 min. L-012 chemiluminescence was expressed as relative light units per milligram of dry tissue weight per minute. NADPH oxidase activity was quantified from the absorbance with or without NADPH.
Measurement of Ca2+-dependent NO synthase activity
Protein samples were extracted from the aortas after 1 week of Ang II infusion. NO synthase (NOS) activity was measured using the conversion rate of radio-labeled L-[3H] citrulline from L-[3H] arginine (Amersham, Little Chalfont, UK). 21 Duplicate incubations were performed for 60 min at room temperature for each sample in the presence or absence of ethylene glycol tetraacetic acid (2 mmol/l) to determine the Ca2+-dependent NOS activity.
Statistical analysis
All data are expressed as means ± SE. Mean values were compared using analysis of variance. If a statistically significant effect was found, Scheffe’s F test was performed to detect the difference between groups. Values of p<0.05 were considered statistically significant.
Results
Expression of AT2 in the aortas of AT2-Tg/apoE-/- mice
We generated five different AT2-Tg lines and then crossed AT2-Tg901 and AT2-Tg902 mice, which expressed aortic AT2 most abundantly, with apoE-/- mice. As the findings obtained from AT2-Tg901/apoE-/- and AT2-Tg902/apoE-/- mice were similar, we here described the result from AT2-Tg901/apoE-/- that expressed aortic AT2 (10.1 ± 1.8 fmol/mg protein) (Supplemental Figure 1) more abundantly than AT2-Tg902/apoE-/- mice (8.9 ± 1.2 fmol/mg protein). The proportion of aortic AT2 relative to AT1 was 33% ± 0.1%, identical to that in AT2-Tg901 mice aortas (39% ± 0.2 %). AT2 was not detected in the aortas of apoE-/- mice, and there were no significant differences in aortic AT1 receptor density between apoE-/- and AT2-Tg901/apoE-/- mice (Supplemental Figure 1). The Kd value (nmol/l) of AT2 in the aorta from AT2-Tg901/apoE-/- mice was 0.39 ± 0.05, similar to that in AT2-Tg901 (0.31 ± 0.01). These animals had similar weights relative to their control littermates (designated as apoE-/- mice), and there was no evidence of obvious morphological changes of the aorta.
Vascular AT2 activation inhibits atherosclerosis development in apoE-/- mice
We first compared the atherosclerotic lesion area between apoE-/- and AT2-Tg/apoE-/- mice fed a high-cholesterol diet for 8 weeks; however, no significant difference was observed between groups. We next examined the effect of a subpressor dose of Ang II (100 ng/kg/min) on atherosclerotic lesion development. Atherosclerotic lesion area was markedly increased in apoE-/- mice (from 3.0% ± 0.4% to 14.5% ± 1.6%, p<0.001); however, Ang II-induced atherosclerotic lesion development was substantially reduced by 53% in AT2-Tg/apoE-/- mice (p<0.05) (Figures 1(a) and 1(b)), suggesting that activation of the vascular AT2 exerts anti-atherogenic actions. Hemodynamic parameters and lipid profiles did not differ between the two groups (Supplemental Figures 2A and 2B).

Vascular angiotensin II (Ang II) type 2 receptor (AT2) inhibits Ang II-induced atherosclerotic lesion formation. (A) Representative photographs of en face Oil Red O-stained aortas in apoE-deficient (apoE-/-) and AT2 transgenic (AT2-Tg)/apoE-/- (AT2-Tg/apoE-/-) mice treated with phosphate-buffered saline (PBS) or Ang II (100 ng/kg/min). The scale bar shows 3 mm intervals. (B) Quantitative analysis showing a significant reduction in the percentage of lesion coverage in the aorta in Ang II-treated AT2-Tg/apoE-/- mice. Values are the mean ± SE for at least 8 mice in each group. *p<0.01 vs. PBS-treated apoE-/- mice. #p<0.05 vs. PBS-treated AT2-Tg/apoE-/- mice. †p<0.05 vs. Ang II-treated apoE-/- mice.
Blockade of the bradykinin/NO system by L-NAME or icatibant abolishes AT2-mediated anti-atherogenic actions
We previously demonstrated that targeted overexpression of the AT2 in VSMCs exerted an antipressor response to Ang II through a kinin/NO-dependent mechanism. 16 To elucidate the involvement of the kinin/NO system in AT2-mediated anti-atherogenic actions, L-NAME or icatibant was administered in combination with the Ang II infusion. Co-treatment with L-NAME did not affect the Ang II-induced atherosclerotic lesions in apoE-/- mice; however, the lesion area in L-NAME-treated AT2-Tg/apoE-/- mice was significantly increased to a similar extent as that seen in apoE-/- mice (13.6% ± 1.0% vs. 12.1% ± 1.6%, apoE-/- vs. AT2-Tg/apoE-/-; p=n.s.) (Figures 2(a) and 2(b)). Similarly, the AT2-mediated anti-atherogenic action observed in AT2-Tg/apoE-/- mice was completely reversed by icatibant treatment. Considering that the bradykinin type 2 receptor is exclusively expressed in the endothelium, these results suggest that the endothelial bradykinin/NO system is likely to be involved in AT2-mediated anti-atherogenic action.
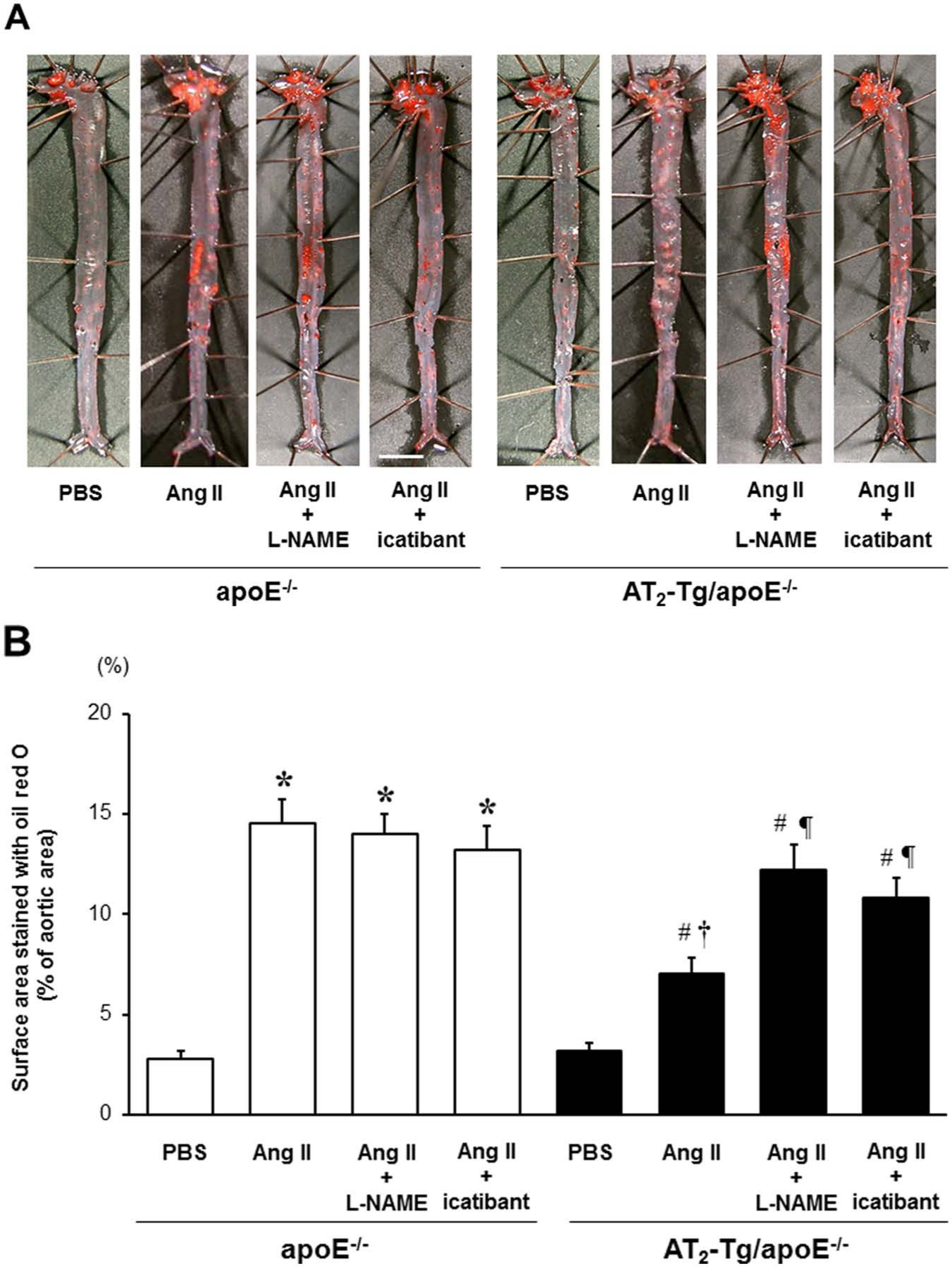
L-NG-nitroarginine methyl ester (L-NAME) or icatibant abolishes vascular angiotensin II type 2 receptor (AT2)-mediated anti-atherogenic effects. (a) Representative photographs of en face Oil Red O-stained aortas in apoE-/- and AT2 transgenic/apoE-/- (AT2-Tg/ apoE-/-) mice treated with L-NAME or icatibant in parallel with Ang II infusion. L-NAME (1 mg/ml) was added in the drinking water of mice in parallel with Ang II (100 ng/kg/min). Icatibant (70 µg/kg per day) was co-infused with Ang II using an osmotic pump. The scale bar shows 3 mm intervals. (b) Quantitative analysis showing a significant reduction of Ang II-induced atherosclerotic lesions in AT2-Tg/apoE-/- mice, a phenomenon that was completely reversed by L-NAME or icatibant treatment. Values are the mean ± SE for at least 8 mice in each group. *p<0.05 vs. phosphate-buffered saline (PBS)-treated apoE-/-. #p<0.05 vs. PBS-treated AT2-Tg/apoE-/- mice. †p<0.05 vs. Ang II-treated apoE-/- mice. ¶p<0.05 vs. Ang II-treated AT2-Tg/apoE-/- mice.
Vascular AT2 activation increases Ca2+-dependent NOS activity in apoE-/- mice
To further elucidate the activation of an endothelial bradykinin/NO system in AT2-Tg/apoE-/- mice, Ca2+-dependent NOS activity in the aorta was examined after 1 week of Ang II infusion, in which few adhesive mononuclear cells were observed in the intima (data not shown). In the absence of Ang II infusion, Ca2+-dependent NOS activity did not differ between the two groups of mice (Figure 3). In contrast, Ang II infusion into AT2-Tg/apoE-/- mice markedly increased Ca2+-dependent NOS activity (67% increase vs. PBS infusion, p<0.01), but had no effect on that in apoE-/- mice. This finding further supports the notion that the AT2-mediated enhancement of NOS activation and the subsequent NO production through an endothelial kinin/NO-dependent mechanism play a critical role in AT2-mediated anti-atherogenic action.
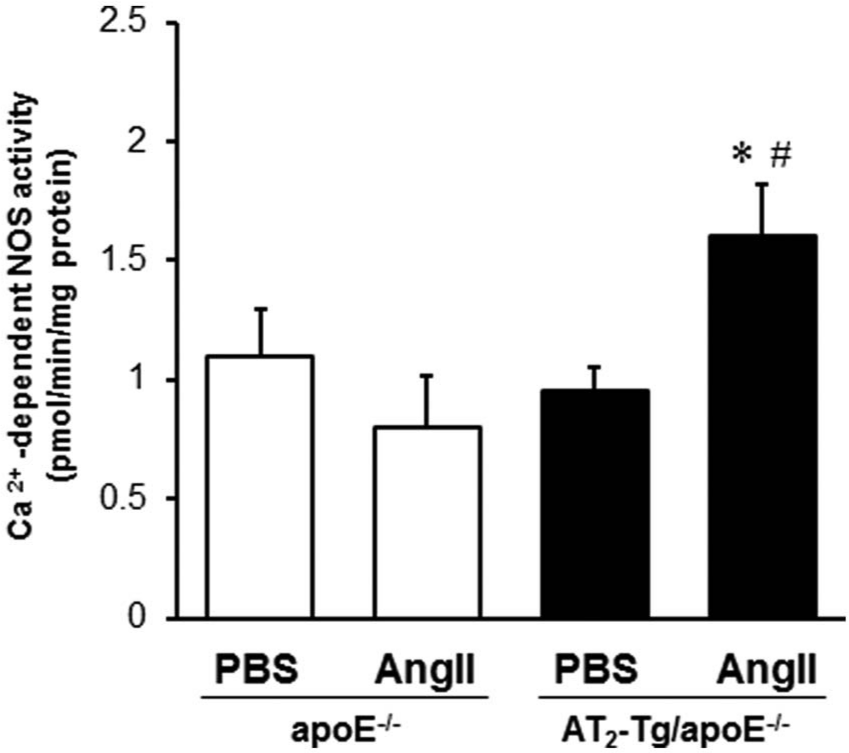
Vascular angiotensin II type 2 receptor (AT2) activation increases Ca2+-dependent nitric oxide synthase (NOS) activity in apoE-/- mice. One week of Ang II infusion significantly enhanced Ca2+-dependent NOS activity in the aortas of angiotensin II type 2 receptor (AT2) transgenic/apoE-/- (AT2-Tg/apoE-/-) mice. The bar graphs represent the mean ± SE of the conversion rate from [3H]-arginine to [3H]-citrulline (pmol/min/mg protein). At least 4 mice were tested in each group. *p<0.05 vs. vessels from phosphate-buffered saline (PBS)-treated AT2-Tg/apoE-/- mice. #p<0.01 vs. vessels from Ang II-treated apoE-/- mice.
Vascular AT2 inhibits Ang II-induced VCAM-1 expression and monocyte/macrophage accumulation
In the early stage of atherosclerosis, activated endothelial cells expressing various kinds of adhesion molecules such as VCAM-1 play a critical role in the accumulation of mononuclear cells within the intima. 22 Since endothelium-derived NO has been shown to inhibit adhesion molecule expression, 23 we examined VCAM-1 expression levels and monocyte/macrophage accumulation by immunochemistry after 2 weeks of Ang II infusion. In the absence of Ang II, the VCAM-1 expression was equivalent between the two groups of mice. Consistent with the previous data, 24 Ang II infusion significantly increased the expression of VCAM-1 on the inner surface and medial wall in apoE-/- mice, whereas it was markedly inhibited in AT2-Tg/apoE-/- mice (Figures 4(a) and 4(b)). Similarly, Ang II-induced monocyte/macrophage accumulation in the luminal surface was significantly decreased in AT2-Tg/apoE-/- mice compared with apoE-/- mice (Figures 4(c) and 4(d)), suggesting that augmented NO production inhibits VCAM-1 expression, resulting in attenuated accumulation of monocytes/macrophages in AT2-Tg/apoE-/- mice.

Vascular angiotensin II type 2 receptor (AT2) inhibits Ang II-induced expression of vascular cell adhesion molecule-1 (VCAM-1) and monocyte/macrophage accumulations. (A) Representative positive VCAM-1 stained aortic arch sections from mice treated with 2 weeks of Ang II infusion (magnification ×200; insets, magnification ×400). L: lumen, M: media. (B) Quantitative analysis showing a significant increase in Ang II-induced expression of VCAM-1 in both the intimal and medial layers of apoE-/- mice. The same was markedly suppressed in AT2 transgenic/apoE-/- (AT2-Tg/apoE-/-) mice. Values are the mean ± SE for at least 6 mice in each group. *p<0.01 vs. vessels from phosphate-buffered saline (PBS)-treated apoE-/- mice. #p<0.01 vs. vessels from Ang II-treated apoE-/- mice. (C) Representative monocyte/macrophage antibody-2 (MOMA-2) positive-stained aortic arch sections from mice treated with 2 weeks of Ang II infusion (magnification ×100, and insets, magnification ×300). L: lumen, M: media. (D) Quantitative analysis showing a significant increase in Ang II-induced accumulation of MOMA-2 positive cells attached to the intimal surfaces in apoE-/- mice but suppression in AT2-Tg/apoE-/- mice. Values are the mean ± SE for at least 6 mice in each group. *p<0.05 vs. vessels from PBS-treated apoE-/- mice. #p<0.01 vs. vessels from Ang II-treated apoE-/- mice.
We also examined the effects of L-NAME on Ang II-induced VCAM-1 expression and monocyte/macrophage accumulation. L-NAME treatment further increased Ang II-induced VCAM-1 expression in both apoE-/- and AT2-Tg/apoE-/- mice; however, the extent was much larger in AT2-Tg/apoE-/- mice than in apoE-/- mice. Consequently, there was no significant difference in VCAM-1-positive area between apoE-/- and AT2-Tg/apoE-/- mice after L-NAME treatment (Supplemental Figure 3A). Similarly, L-NAME treatment markedly augmented Ang II-induced monocyte/macrophage accumulation in AT2-Tg/apoE-/- mice to the same extent as that in apoE-/- mice (Supplemental Figures 3B and 3C). These findings further supported the current hypothesis that vascular AT2 overexpression exerts anti-atherogenic action through an endothelial kinin/NO system.
Vascular AT2 attenuates Ang II-induced oxidative stress by inhibiting the accumulation of superoxide-producing mononuclear leukocytes
To elucidate the manner in which AT2 counteracts AT1-mediated oxidative stress, cell-specific generation of superoxide anions was examined. In the absence of Ang II, the fluorescence intensity of DHE-positive mononuclear cells in the intima did not differ between apoE-/- and AT2-Tg/apoE-/- mice (Figures 5(a) and 5(b)). In contrast, 2 weeks of Ang II infusion into apoE-/- mice significantly increased the fluorescence intensity, particularly in the intima, which was completely inhibited in AT2-Tg/apoE-/- mice. Consistent with this finding, the mRNA expression level of Nox2, phagocytic NADPH oxidase, 25 was markedly increased in apoE-/- mice (5.1-fold vs. PBS; p<0.05) after Ang II infusion (Supplemental Figure 4A), but was significantly reduced in AT2-Tg/apoE-/- mice, suggesting that AT2-mediated anti-oxidative action is mostly exerted by inhibiting the accumulation of superoxide-producing mononuclear leukocytes.

Vascular angiotensin II type 2 receptor (AT2) attenuates Ang II-induced oxidative stress by inhibiting the accumulation of superoxide-producing mononuclear leukocytes. (A) Representative photographs showing dihydroethidium (DHE)-positive mononuclear cells attached to the intimal layer (magnification ×600). Arrows indicate DHE-positive mononuclear cells attached to the intimal layer. M: media, L: lumen. (B) Quantitative analysis showing Ang II-induced augmentation of superoxide production in apoE-deficient (apoE-/-) mice, a phenomenon that was profoundly attenuated in AT2 transgenic/apoE-/- (AT2-Tg/apoE-/-) mice. Superoxide levels were calculated as the ratio (fold increase) to the control values obtained from sequential unstained sections from each mouse. The bar graphs represent the mean ± SE relative to phosphate-buffered saline (PBS)-treated apoE-/- mice set at 100%. At least 6 mice were tested in each group. *p<0.05 vs. vessels from PBS-treated apoE-/- mice. #p<0.05 vs. vessels from Ang II-treated apoE-/- mice.
We also examined the activity of NADPH oxidase after 1 week of Ang II infusion, at which time few adhesive mononuclear cells were observed in the intima (data not shown), and found no significant difference between the two groups of mice (Supplemental Figure 4B). These findings suggest that vascular-targeted overexpression of AT2 exerts its anti-oxidative action by attenuating the accumulation of superoxide-producing mononuclear leukocytes rather than by a direct effect on vascular NADPH oxidase activity.
Discussion
This is the first study to indicate that targeted overexpression of AT2 in VSMCs significantly attenuates atherosclerotic lesion development in an endothelial kinin/NO-dependent manner. We previously demonstrated that AT2 stimulation in VSMCs promotes intracellular acidosis by blocking the amiloride-sensitive Na+/H+ exchanger, leading to activation of kininogenases followed by bradykinin release, which causes endothelial NO-dependent vasodilation in mice. 16 Accumulating evidence supports the notion that the AT2 stimulates a vasodilator signaling cascade that includes bradykinin, NO, and guanosine cyclic 3’, 5’-monophosphate. 26 The present study using AT2-Tg/apoE-/- mice provides new evidence that the AT2 bradykinin NO system exerts its anti-atherogenic action by attenuating the increased VCAM-1 expression and monocyte/macrophage accumulation, both of which are essentially involved in the early stage of atherosclerosis development. Furthermore, the protective effect of AT2 on oxidative stress was mostly exerted through reduced accumulation of superoxide-producing mononuclear leukocytes rather than by a direct effect on vascular NADPH oxidase activity. These findings provide new insight into the AT2-mediated anti-atherogenic actions and support the possibility that direct AT2 activation could be a potential therapeutic strategy for preventing the development of atherosclerotic cardiovascular diseases.
AT2 is substantially expressed by endothelial cells of the microvasculature,27,28 whereas its expression in larger arteries such as the thoracic aorta was barely detectable by a conventional binding assay or immunohistochemistry.16,28 However, an autoradiography study revealed that a low level of aortic AT2 expression was detected in the cell layer at the medial–adventitial border rather than the medial–endothelial wall. 29 Similarly, using a newly developed polyclonal anti-AT2 antiserum, Utsunomiya et al. 14 reported that AT2 expression was observed only in the aortic muscular medial layer and not in the endothelium. Furthermore, both AT1 and AT2 expression were up-regulated in the aortas of apoE-/- mice fed an atherogenic diet.9,15 Sales et al. showed that the mRNA expression of the AT2 was threefold higher in apoE-/- mice fed an atherogenic diet compared with those on standard chow diet. 15 Iwai et al. also observed that the AT2 was expressed in both the lesion and the aortic medial wall in apoE-/- mice on a high-cholesterol diet. 9 The previous in vitro studies investigating the biological effects of AT2 in VSMCs and endothelial cells showed the discordant results, which is probably due to the difference in animal species and cell lines to be tested. The biological relevance of these results should be assessed in vivo experiments. From this point of view, AT2-Tg/apoE-/- mice in which AT2 is overexpressed in aortic VSMCs might be a suitable model for investing the vascular action of aortic AT2 in atherogenesis. Our findings suggest that AT2 receptor in VSMCs plays a critical role in atherogenesis, especially in hypercholesterolemia which elicits re-expression of the aortic AT2 in the medial cell layer including VSMCs.
Studies investigating the effect of AT2 on the development of atherosclerosis showed conflicting results. AT2 exerted an anti-atherogenic effect in AT2/apoE double-deficient mice, 9 whereas no such action was observed in AT2/LDL double-deficient mice. 15 These discrepancies most likely resulted from the different stages of atherosclerosis investigated. For example, the atherosclerotic lesion areas in AT2/apoE double-deficient mice were estimated after 10 weeks on a high-cholesterol diet, corresponding to the early stage of atherosclerosis, whereas the AT2/LDL double-deficient mice were analyzed at 5 months of age, at which point the plaque evolution was further advanced. Similarly, Daugherty et al. demonstrated that AT2 antagonist PD123319 treatment exaggerated the Ang II-induced atherosclerotic lesion area in 2-month-old apoE-/- mice, whereas no such effect was observed at the age of 11 months. 7
Long-term treatment with a Western diet has been shown to impair endothelial function in apoE-/- mice.30,31 Considering that Ang II-induced Ca2+-dependent NOS activity was enhanced in AT2-Tg/apoE-/- mice, the AT2-mediated anti-atherogenic effect is likely to be exerted in the early stage of atherosclerosis, during which endothelial function was preserved. Therefore, it is of great importance that the tissue-specific effect and time-dependent action of vascular AT2 is carefully considered for estimating AT2-mediated anti-atherogenic actions in the multifactorial process of atherosclerosis.
Reactive oxygen species (ROS) and NO are crucial for the development of atherosclerosis. 32 AT2 has been shown to both increase NO production 16 and inhibit ROS production 9 ; however, the precise mechanism for AT2-mediated modulation of oxidative stress remains to be determined. Iwai et al. 9 demonstrated that the AT2 blocker PD123319 augmented Ang II-induced NADPH oxidase activity in rat fetal VSMCs, suggesting that the anti-atherogenic effect of AT2 occurs, in part, through the inhibitory effect on NADPH oxidase activation, a dominant source of oxidative stress in the vasculature. 25 However, the present study did not show an inhibitory effect of AT2 on NADPH oxidase activity (Supplemental Figure 4B). Iwai et al. stimulated fetal VSMCs with 10-7 M Ang II and observed a significant increase in NADPH oxidase activity, 9 while the present study showed that a subpressor dose of Ang II did not increase NADPH oxidase activity (Supplemental Figure 4B), suggesting that AT2 stimulation was likely to antagonize AT1-mediated NADPH oxidase activity by directly inhibiting crosstalk of the two receptors. Accordingly, the discrepancy between these two experiments might be attributable to the extent of AT1-mediated augmentation of NADPH oxidase activity.
The clinical relevance of AT2 receptor has not been confirmed because of the lack of a selective AT2 agonist available in clinical practice. Recently, compound 21 (C21), a non-peptide, orally active and selective AT2 agonist has been developed. 10 Direct AT2 stimulation by C21 has been reported to exert beneficial effects on cardiac and vascular function in rodents through an increased production of NO and/or suppression of the inflammatory response.33,34 Rehman et al. 34 reported that endothelium-dependent relaxation in isolated rat aortas was enhanced in the C21–losartan combination groups compared with losartan alone, suggesting that C21 treatment increases NO production, ARB treatment inhibits superoxide production, and both lead to the increased NO bioavailability. Considering that the expression of AT2 receptor is up-regulated in various pathological conditions including post-myocardial infarction, heart failure, and arterial injury,11–13 the beneficial effect of AT2 is expected to be assessed in the future clinical trials. Our findings strongly support the notion that direct AT2 stimulation exerts anti-atherogenic action and could be a potential therapeutic strategy for producing a clinically beneficial effect on the cardiovascular system.
Conclusions
AT2 activation in VSMCs markedly inhibited the initiation of atherosclerotic lesion development in an endothelial bradykinin/NO-dependent manner, and AT2-mediated modulation of oxidative stress was mostly attributable to the enhanced NO production and inhibited accumulation of superoxide-producing leukocytes. These findings provide novel evidence of vascular AT2-mediated anti-atherogenic actions and new insight into the mechanism for AT2-mediated anti-oxidative action, which could resolve the controversial issue regarding the AT2-mediated effect on atherosclerosis.
Footnotes
Conflict of interest
None declared.
Funding
This work was supported by a grant from the Ministry of Education, Culture, Sports, Science and Technology of Japan.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.