
Abstract
Introduction:
Vascular smooth muscle cells (VSMCs) are essential for maintaining vasculature homeostasis and function. By influence on its growth and activation both proinflammatory cytokines and peptides of the renin–angiotensin system (RAS) are potent regulators of VSMCs. Interestingly, angiotensin (Ang) II and Ang-(1–7) elicit opposite effects on VSMC activation, differentiation and proliferation. It has been suggested that statins, besides anti-inflammatory effects, may also modulate VSMC activation by their influence on the RAS.
Methods:
The effect of atorvastatin on Ang I metabolism in a culture of explanted rat VSMCs was examined by liquid chromatography-mass spectrometry (LC-MS); expression of mRNA of the main RAS enzymes in VSMC was assessed by real-time polymerase chain reaction (PCR).
Results:
In VSMC culture Ang-(1–7) was identified as a major product of Ang I metabolism. In this setting, TNF-α (1 ng/ml) caused a decrease in the conversion of Ang I to Ang-(1–7). This effect was accompanied by a decrease of mRNA expression of neutral endopeptidase (NEP) and angiotensin converting enzyme 2 (ACE2) and increase of mRNA of ACE. Interestingly, atorvastatin (3 μM) attenuated the effects of TNF-α on Ang-(1–7) production as well as reversed the influence of TNF-α on ACE and ACE2 expression.
Conclusions:
Enhancement by atorvastatin of the ACE2/Ang-(1–7) axis in VSMCs could represent a new and beneficial mechanism on cardiovascular action of this widely used drug.
Introduction
Vascular smooth muscle cells (VSMCs) are essential for maintaining vascular homeostasis and function. 1 Their increased proliferation and migration upon stimulation by proinflammatory cytokines (e.g. TNF-α), alongside enhanced synthesis and secretion of extracellular matrix proteins (e.g. growth factors, chemokines, cytokines) all contribute to vascular remodeling and inflammation which are recognized hallmarks of hypertension and atherosclerosis.2,3
The depiction of the renin–angiotensin system (RAS) has changed over recent years. The ‘new’ peptides (e.g. angiotensin (Ang)-(1–7)) and enzymes (e.g. angiotensin converting enzyme 2, (ACE2)) have been described and Figure 1 highlights the main pathways of angiotensin metabolism.4,5 It has also become clear that angiotensins can be generated not only in systemic circulation, but also locally, in several tissues and organs. The RAS is one of the most potent regulators of VSMC growth and function.4,6,7 It has been well documented that Ang II is a strong inducer of VSMC proliferation by activating the AT1 receptor (AT1R) and ERK1/2 and p38 MAP kinases7 –9 and it also plays an important role in vascular calcification. 10 Recently, increased Ang II-induced aortic remodeling was demonstrated in ACE2 knockout mice. 11 However, data about the action as well as local production of ‘new’ Ang peptides in VSMC are scarce.

Schematic depiction of the main pathways of angiotensin (Ang) metabolism. ACE: angiotensin converting enzyme; AT1: Ang II type 1 receptor; AT2: Ang II type 2 receptor; NEP: neutral endopeptidase.
HMG-CoA reductase inhibitors (statins) act on VSMCs in a complex, pleiotropic manner, influencing their migration, proliferation, differentiation and survivall.12 –15 Intriguingly, statins may interfere in the local RAS, resulting in regulation of VSMC activation, although the mechanism of their action remains unclear. At least partly, statins may act on the level of intracellular signaling as it has been reported that atorvastatin may reduce Ang II- and TNF-α-elicited induction of NF-κB, which mediates the migration and proliferation of VSMCs.7,16 –18 However, there is lack of information as to whether statins may directly influence function of RAS in VSMCs.
The aim of the study was to examine the effect of atorvastatin on Ang I metabolism and the expression of the main enzymes of the RAS in a culture of resting and TNF-α-stimulated rat VSMCs.
Materials and methods
Cell culture
All procedures were approved by an Ethical Committee of the School of Medicine, Jagiellonian University.
Primary cultures of Wistar rat aortic vascular smooth muscle cells were grown according to the explant outgrowth technique as previously described. 19 Cells were cultured in Dulbecco’s modified Eagle’s medium mixed with F-12 medium (1:1 v/v) containing 10% fetal calf serum (FCS), 100 U/ml penicillin, 100 μg/ml streptomycin, amphotericin B (0.25 μg/ml). The cultured cells showed the characteristic ‘hills and valleys’ growth pattern and α-SM-actin positive staining by indirect immunocytochemistry: the cells were lacking von Willebrand Factor immunoreactivity. Cells in passages 2–4 were grown to confluence, and then were exposed to one of the following treatments: TNF-α (1 ng/ml), atorvastatin (3 µM), and atorvastatin (3 µM) + TNF-α (1 ng/ml). Statin concentration had been chosen based on the activity/toxicity profile of the drug determined in our preliminary experiments. Resting VSMCs in passages 2–4 grown for 24 h alongside the exposed cultures served as the control group in the experiment. Two aortas were used to establish one primary cell culture line which was used in a single experiment. Three experimental repetitions were performed. All experiments were performed in triplicate.
After 24 h of incubation culture, the medium was removed and the cells were washed with phosphate buffered saline (PBS) buffer. Then the cells were incubated in 500 µl of PBS buffer containing 1 μM Ang I for 15 min. After that, the supernatant was acidified and subjected to liquid chromatography-mass spectrometry (LC-MS) analysis and the cells were harvested for the real-time PCR analysis.
RNA extraction, cDNA synthesis, and real-time PCR
Total RNA was extracted by using TRIzol® Reagent (Gibco-BRL, USA) according to manufacturer’s instructions. The cDNA was synthesized by the reverse transcription of 1.5 µg of total RNA from each sample using oligo(dT)18 primer (Fermentas, Canada) and M-MuLV reverse transcriptase (200 U/µl) (Fermentas, Canada) in a total volume of 10 µl as previously described. 20 The mRNA levels were determined by quantitative real-time PCR performed with the 7900HT Fast Real-Time PCR System (Applied Biosystems, USA), using the SYBR® Green JumpStart™ Taq ReadyMix ™ for quantitative PCR (Sigma, USA) as previously described. 20 Levels of ACE, ACE-2 and NEP mRNAs were normalized to GAPDH mRNA. Primers for rat ACE: 5’- ggg cag tgg cta cga gca tg -3’ and 5’- agt tct cct ggt gat gct tc -3’, ACE-2: 5’- acc ctt ctt aca tca gcc cta ctg -3’ and 5’- tgt cca aaa cct acc cca cat at -3’, and NEP: 5’- gca gaa atc aga tcg tct tcc ccg c -3’ and 5’- ctg agt cca cca gtc aac gag gt -3’ were designed with Primer BLAST software (http://www.ncbi.nlm.nih.gov/tools/primer-blast) taking into account primer dimer, self-priming formation and primer melting temperature, and primers for GAPDH: 5’- tga ttc tac cca cgg caa gt -3’ and 5’- agc atc acc cca ttt gat gt -3’ were used. 21
Sample preparation for LC-MS measurements
Samples were purified and concentrated using Ultra-Micro Spin C-18 column (Harvard Apparatus, USA) as described previously. 22 Briefly, 400 μl of sample were applied on columns and centrifuged (30 s, 1000 x g). Then columns were washed with 300 μl of 0.1% trifluoroacetic acid (TFA) by centrifugation as above. Finally Ang peptides were eluted by centrifugation with 300 μl of 0.1% TFA in 40% acetonitrile. Then the samples were lyophilized overnight, and dry remainings were reconstituted in 0.1 % TFA for LC-MS analysis. Samples for calibration curves of each of the examined peptides (mixture of standards) were prepared in the same mode as above.
Measurement of Ang peptide concentration by LC-MS method
Separation of Ang peptides was performed on a reversed-phase, high performance liquid chromatography (HPLC) system Ultimate 3000 (Dionex, USA) as described previously.22,23 The column used for separation was Acclaim PepMap 100 C18 column (150 mm x 300 μm ID; 5 μm particle size) with a trap column (5 mm x 1 mm; 5 μm particle size) working at a flow rate of 5 μl/min. Mass spectrometric detection was performed using a LCQ Ion-Trap Mass Spectrometer (Finnigan, San Jose, USA). For Ang peptide detection, selected ion monitoring (SIM) mode was used, set at 450.32, 466.38, 523.95, 592.39, 648.95, 665.27 and 775.27 Da for Ang-(1–7), Ang III, Ang II, Ang-(1–9), Ang I, Ang-(1–5) and Ang IV, respectively. Acquired data were analyzed by Xcalibur Software, version 2.0. Concentrations of Ang peptides were calculated using the standard calibration curves, constructed by linear regression analysis plotting of peak area versus peptide concentration. Calibration curves were prepared for each examined peptide at a concentration range of 2.5–250 ng/ml.
Chemicals
Ang I, Ang III, Ang IV and Ang-(1-9) and Ang-(1-5) were purchased from Bachem (USA). Ang II and Ang-(1-7) were purchased from Sigma Chemicals (USA). Formic acid (99%), TFA and ammonium formate were purchased from Fluka (USA). Acetonitrile (JT Baker, USA), and ddH2O (Simplicity UV, Millipore) were HPLC grade.
Statistics
Concentrations of angiotensins were expressed as pg/µl/mg of protein. All values in the figures and text are expressed as mean±standard error of the mean (sem) of n observations. One-way analysis of variance (ANOVA) followed by Tukey’s post-hoc test was used for analysis of the data (STATISTICA PL 10.0, StatSoft, Poland). A value of p<0.05 was considered statistically significant.
Results
Incubation of VSMCs with TNF-α resulted in a decrease of expression of mRNA of NEP and ACE2 and an increase in expression of mRNA of ACE (Figure 2) as compared to the resting cells transcript levels (control group). In turn, incubation of VSMCs with atorvastatin resulted in the decrease of expression of mRNA of ACE and NEP (Figure 2) as compared to the control group. Interestingly, atorvastatin reversed the changes in ACE and ACE2 mRNA expression caused by TNF-α (Figure 2). It is noteworthy that atorvastatin and TNF-α synergistically downregulated the levels of NEP mRNA as compared to the resting cells (Figure 2).
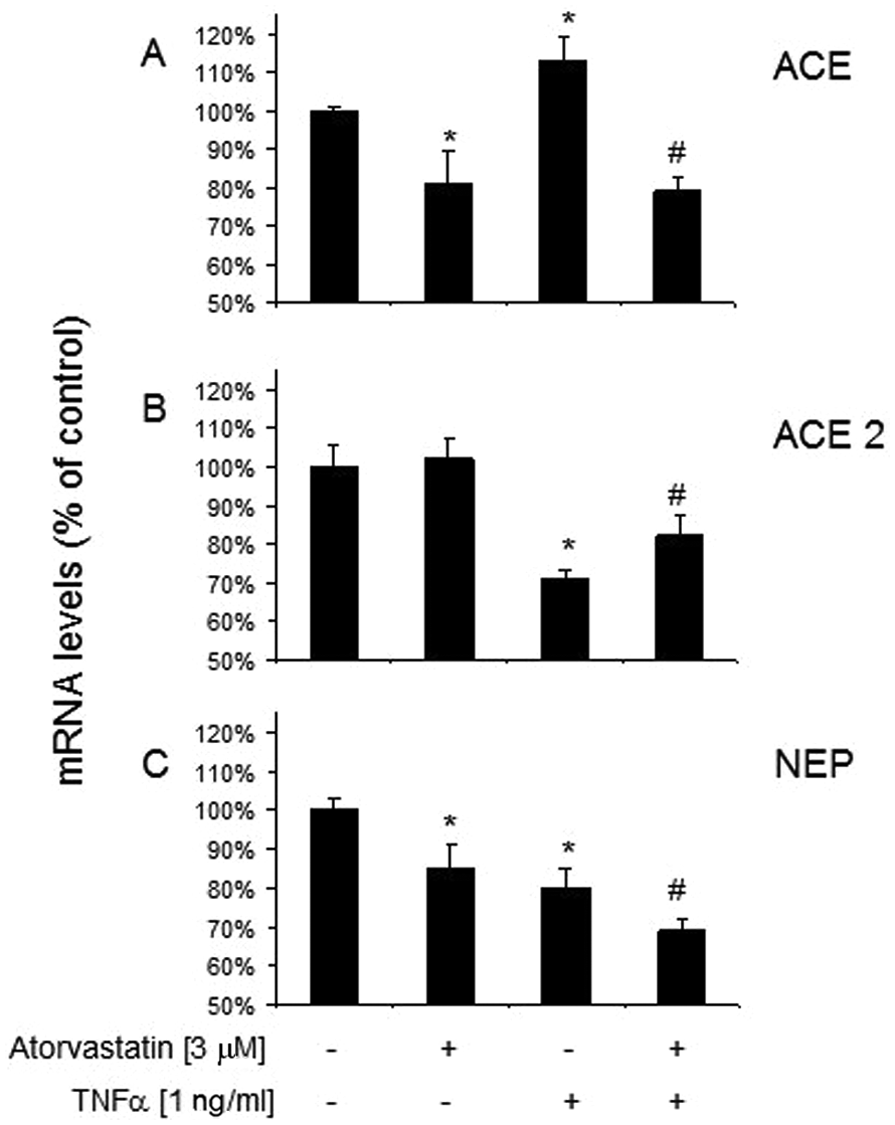
Changes in expression of (A) angiotensin converting enzyme (ACE), (B) ACE2 and (C) neutral endopeptidase (NEP) in vascular smooth muscle cells (VSMCs) treated by TNF-α and/or atorvastatin. Transcript levels were assessed by real-time polymerase chain reaction (PCR) after 24 h exposure to TNF-α (1 ng/ml), atorvastatin (3 µM), and atorvastatin (3 µM) + TNF-α (1 ng/ml). Significance of differences between groups was established by one-way analysis of variance (ANOVA) followed by Tukey’s post-hoc test. n=3 experiments (done in triplicates). *p<0.05 vs resting cells; # p<0.05 vs TNF-α-treated group.
In VSMC culture, Ang I was mainly converted to Ang-(1–7) (Figure 3). Metabolism of Ang I resulted also in formation of Ang-(1–9), Ang-(1–5) and Ang II: notably their concentrations were 6.7, 50 and 52-fold, respectively, lower than the concentration of Ang-(1–7) (Figure 3). Incubation of VSMCs with TNF-α caused a substantial decrease in Ang-(1–7) production by VSMCs (Figure 4). Importantly, this effect was significantly attenuated by atorvastatin (Figure 4). Atorvastatin attenuated also the decrease in Ang-(1–9) production in TNF-α-stimulated VSMCs but it did not affect the peptide production in resting cells.

Pattern of angiotensin (Ang) peptides in resting rat vascular smooth muscle cells (VSMCs) incubated with Ang I (1 µM) for 15 min. Data obtained by liquid chromatography-mass spectrometry (LC-MS) measurements were presented as mean±standard error of the mean (sem) of n=3 experiments (done in triplicates)

Influence of TNF-α and/or atorvastatin on the production of angiotensin (Ang) II and Ang-(1–7) by vascular smooth muscle cells (VSMCs). Peptides were produced in 15 min incubations of VSMCs with Ang I (1 µM) as a substrate. Data were presented as mean±standard error of the mean (sem) of n=3 experiments (done in triplicates). Significance of differences between groups was established by one-way analysis of variance (ANOVA) followed by Tukey’s post-hoc test. * p<0.05 vs resting cells; # p<0.05 vs TNF-α-treated group.
The amounts of Ang II generated out of Ang I by TNF-α-treated VSMCs did not differ significantly when compared to resting cells (Figure 4).
Discussion
It is well recognized that a variety of cells express RAS components, although the profiles of expression of the main RAS enzymes differ between tissues, yielding different local patterns of products of Ang I conversion.4,22 –25 Regarding VSMCs, the majority of reports deal with Ang II action. 9 Surprisingly, here we identified Ang-(1–7) as a major product of Ang I metabolism both in resting and TNF-α-treated rat VSMCs. Our result may point to the important autocrine/paracrine action of Ang-(1–7) on smooth muscle cells within the vessel wall. Importantly, the ‘new’ ACE2/Ang-(1–7)/Mas receptor axis has been shown to counterbalance the detrimental action of the ‘classical’ ACE/Ang II/AT1R pathway in the cardiovascular system.26,27 Recent findings indicate, that Ang-(1–7) can directly (by Mas receptor activation) and indirectly (via prostacyclin) contribute to ensure cardiovasculature homeostasis.6,28,29 Thus, not only AT1R blockers but also Ang-(1–7) peptidomimetic Mas receptor agonist AVE0991 have recently been shown to have protective, antiproliferative properties as countermeasures to vascular remodeling.26,29,30 It is tempting to speculate that VSMC Ang-(1–7) generation may represent an endogenous protective mechanism within the vessel wall: however, taking into account the differences in VSMC phenotype between in vitro and in vivo settings, this attractive hypothesis requires further investigation.
In our study, TNF-α increased ACE and decreased the levels of ACE2 and NEP mRNAs in VSMCs. Importantly, these changes were associated with a substantial decrease in the formation of Ang-(1–7). It was shown that both TNF-α and Ang II, by sharing several intracellular pathways, play a pivotal role in excessive VSMC proliferation and overgrowth, augmenting vascular inflammation and remodeling.26,31 –33 Whether the TNF-α-elicited decrease of Ang-(1–7) formation may represent a new mechanism linking the RAS and inflammation in vessel wall pathology in vivo remains to be tested.
Despite numerous studies relating to the mechanisms of action of statins in various cells in the cardiovasculature, there is surprisingly little information concerning the influence of this group of drugs on the expression/functioning of RAS components. It has been reported that atorvastatin is able to decrease ACE expression in stimulated macrophages and endothelial cells.34,35 Here, for the first time, we showed that atorvastatin decreased ACE and NEP mRNA expression in resting VSMCs without any influence on ACE2 mRNA expression. Moreover, it was able to partially reverse the unfavorable ACE and ACE2 mRNA changes in VSMCs upon TNF-α activation. Importantly, in our setting such actions of atorvastatin were associated with significant promotion of VSMC Ang-(1–7) production.
Both TNF-α and Ang II have been proposed to play an important role in vessel wall injury in atherosclerosis and hypertension.9,31 –33 It has been also documented that statins are able to abolish TNF-α and Ang II-elicited VSMC migration, proliferation and oxidative stress, and diverse mechanisms of such actions have been proposed (e.g. by inhibition of leptin synthesis, normalization of eNOS, Cu/ZnSOD, EcSOD and NADPH oxidase expression, decrease of production of selected growth factors).7,16,36 –39 It may well be that improvement of function of the ACE2/Ang-(1–7)/Mas receptor axis in VSMCs may represent a new mechanism of beneficial action of atorvastatin in the vessel walls. However, validation of such a mechanism in human pathology as well as elucidation of the molecular details of the influence of atorvastatin on VSMCs and the RAS clearly require further studies.
In summary, we identified Ang-(1–7) as a major product of Ang I metabolism in rat VSMCs. Inflammatory stimulation of VSMCs resulted in a decrease in ACE2 and increase of ACE mRNA expression as well as a decrease of formation of Ang-(1–7); such effects were attenuated by atorvastatin. The molecular basis as well as functional meaning of our results require further investigation.
Footnotes
Acknowledgements
The authors are grateful to Renata Budzyńska for technical help with animal experiments.
Funding
This study was supported by NCN grant No. 2011/01/N/NZ4/03752. Aneta Stachowicz acknowledges the financial support from the project Interdisciplinary PhD Studies ‘Molecular sciences for medicine’ (co-financed by the European Social Fund within the Human Capital Operational Programme).
Conflict of interest
None declared.