
Abstract
The results of density functional theory calculations at the APFD/SDD level are detailed herein in order to study the main steps in the α,β-dehydrogenation of aldehydes and cyclic ketones in the presence of an allylpalladium complex catalyst. The mechanism is believed to proceed via an allylpalladium enolate complex (
Introduction
The conversion of saturated ketones and aldehydes to the corresponding α,β-unsaturated compounds is an important synthetic tool. In particular, the introduction of a double bond in the α,β position is of great interest, because this bond can be subsequently modified to provide a wide range of functional groups, thus potentially opening up an easy route to the synthesis of many organic compounds. Between 1982 and 1983, Shimizu and colleagues1–5 reported a methodology for the regioselective dehydrogenation of carbonyl compounds in the presence of an allylpalladium catalyst. Two competitive reactions—namely, allylation and dehydrogenation—were identified and controlled by adjustment of the specific ligand, solvent, and temperature. For instance, acetonitrile at 80°C was found to be the most suitable solvent for the dehydrogenation pathway. In addition, the molar ratio of the palladium and phosphine ligand was found to be crucial, with the dehydrogenation proceeding efficiently at a ratio of less than two, or even in the absence of the phosphine ligand. With respect to the choice of ligand, 1,2-bis(diphenylphosphino)ethane (dppe) was found to be the best choice for obtaining the dehydrogenation product.
More recently (in 2010), Muzart 6 published a detailed review on the preparation of α, β-unsaturated carbonyls from the corresponding saturated compounds and palladium reagents, with both stoichiometric and catalytic methods being explained. Furthermore, in 2018, Huang et al. 7 developed a method for synthesizing α, β-unsaturated cyclic ketones from the zinc enolate derivatives.
A great deal of recent development has been reported in the use of manganese as a cheap and environmentally friendly catalyst for hydrogenation and dehydrogenation reactions. 8 In addition, Maiti et al. have reported the use of a cobalt catalyst to introduce an internal aliphatic olefin during the Heck-type dehydrogenation reaction,9–11 and palladium catalysis for the selective C–H allylation of arenes.12,13 Furthermore, Mishra et al. 14 have published a detailed review of the catalytic allylation of C(sp2)–H bonds with various transition metal catalysts.
A new route for the preparation of α,β-unsaturated aldehydes and cyclic ketones in the presence allyl diethyl phosphate (ADP), THF and Na2CO3, and a palladium catalyst, without using any ligand, has been developed by the present research group. 15 The overall reaction is shown in Scheme 1.
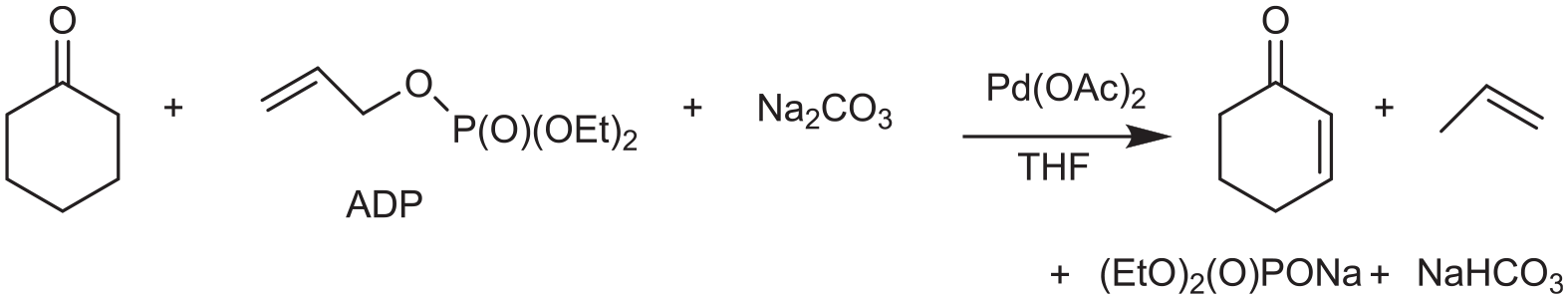
The dehydrogenation of cyclohexanone.
A reasonable mechanism is outlined in Scheme 2. Thus, the reduction of Pd(OAc)2 with cyclohexanone gives Pd0 as the actual catalyst, and the η3-allylpalladium cation is formed in the presence of allyl phosphate. Under basic conditions, enolate is present and reacts with the allylpalladium to give complex
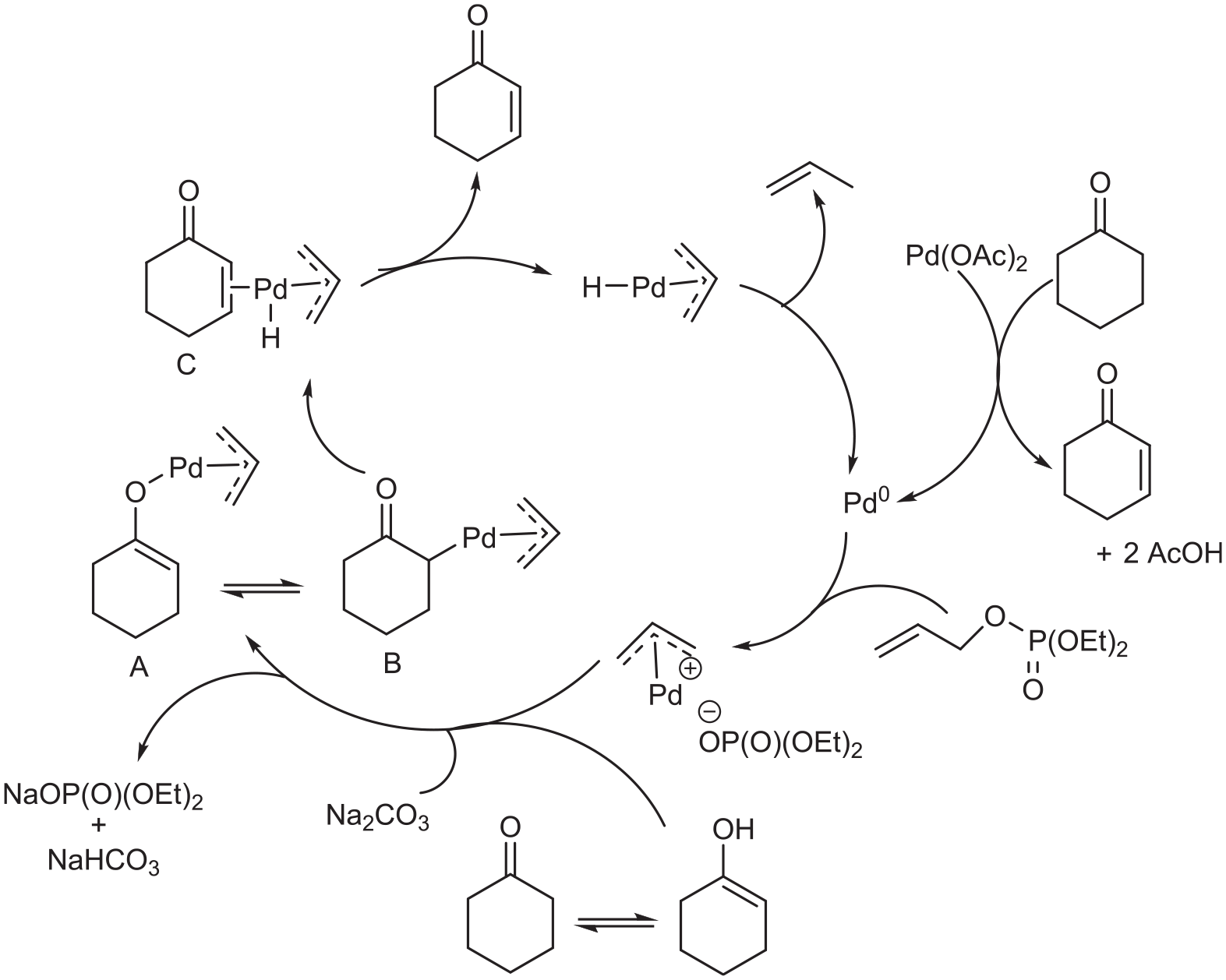
A plausible reaction mechanism for the dehydrogenation of cyclohexanone.
In a versatile computational study on the Tsuji allylation reaction, Keith et al.
16
showed that the observed enantioselectivity in the Pd-catalyzed intramolecular decarboxylative allylic alkylation of allyl enol carbonates is best explained by an inner-sphere pathway. However, to the best of the present authors’ knowledge, no extensive computational studies have been performed on the dehydrogenation reaction in the presence of allylpalladium catalysts. Herein, a computational density functional theory (DFT) investigation of the optimized structures of complexes
Computational methods
All DFT calculations were performed using the Gaussian 16 software 17 supported by the GaussView 6.0 interface. 18 All geometric optimizations and frequency calculations were performed using the APF-D hybrid DFT method including dispersion (keyword APFD) with the SDD basis set. For higher accuracy, the ultrafine grid was used and was specified by the Int = Ultrafine keyword. Frequency calculations were also performed at the same level of theory to identify all of the stationary points as minima (zero imaginary frequencies) or transition states (one imaginary frequency). The transition states were optimized via the synchronous transit-guided quasi-Newton methods (QST2 and QST3). Intrinsic reaction coordinate (IRC) calculations were performed at the same level to verify that the transition states led to the expected reactants and products. Moreover, the calculation with THF as the solvent was accomplished by adding the self-consistent reaction field as SCRF = (SMD, solvent = THF). The discussions herein are based on the Gibbs energies, with the difference in Gibbs energy between the reactant and transition state being used to calculate the energy barrier. All reported energy values refer to the standard conditions, that is, 298.15 K and 1 atm pressure. The calculated imaginary frequencies of all transition-state species, along with the Cartesian coordinates and thermodynamic parameters of all the calculated structures, are provided in the Supplementary Material.
Results and discussion
The optimized geometry of complex
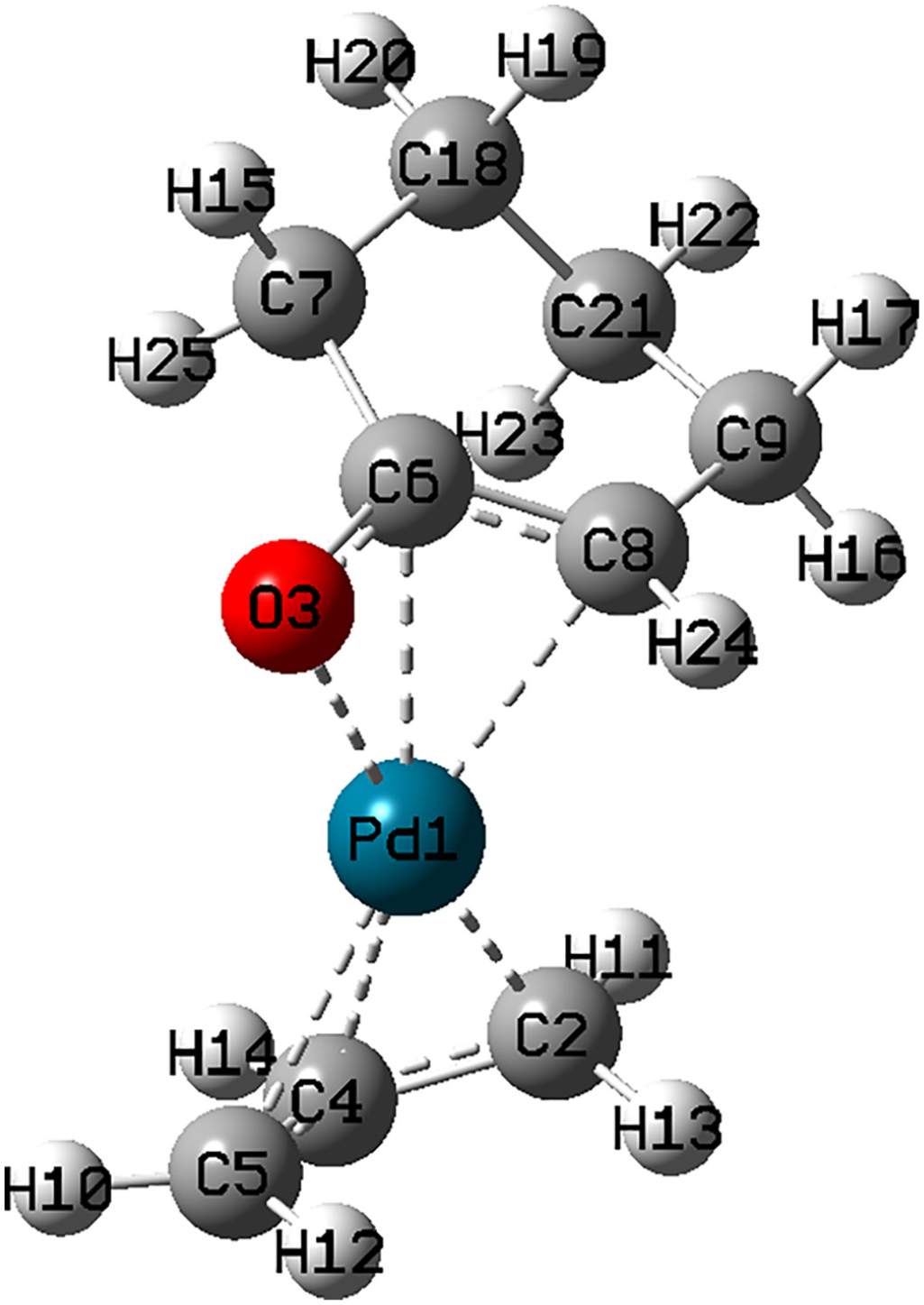
The optimized structure of complex
Selected bond lengths for complex

Complex
The Gibbs energy and the relative Gibbs energies of

The Gibbs energy and the relative Gibbs energies of

These results indicate that complex
The optimized geometry of complex
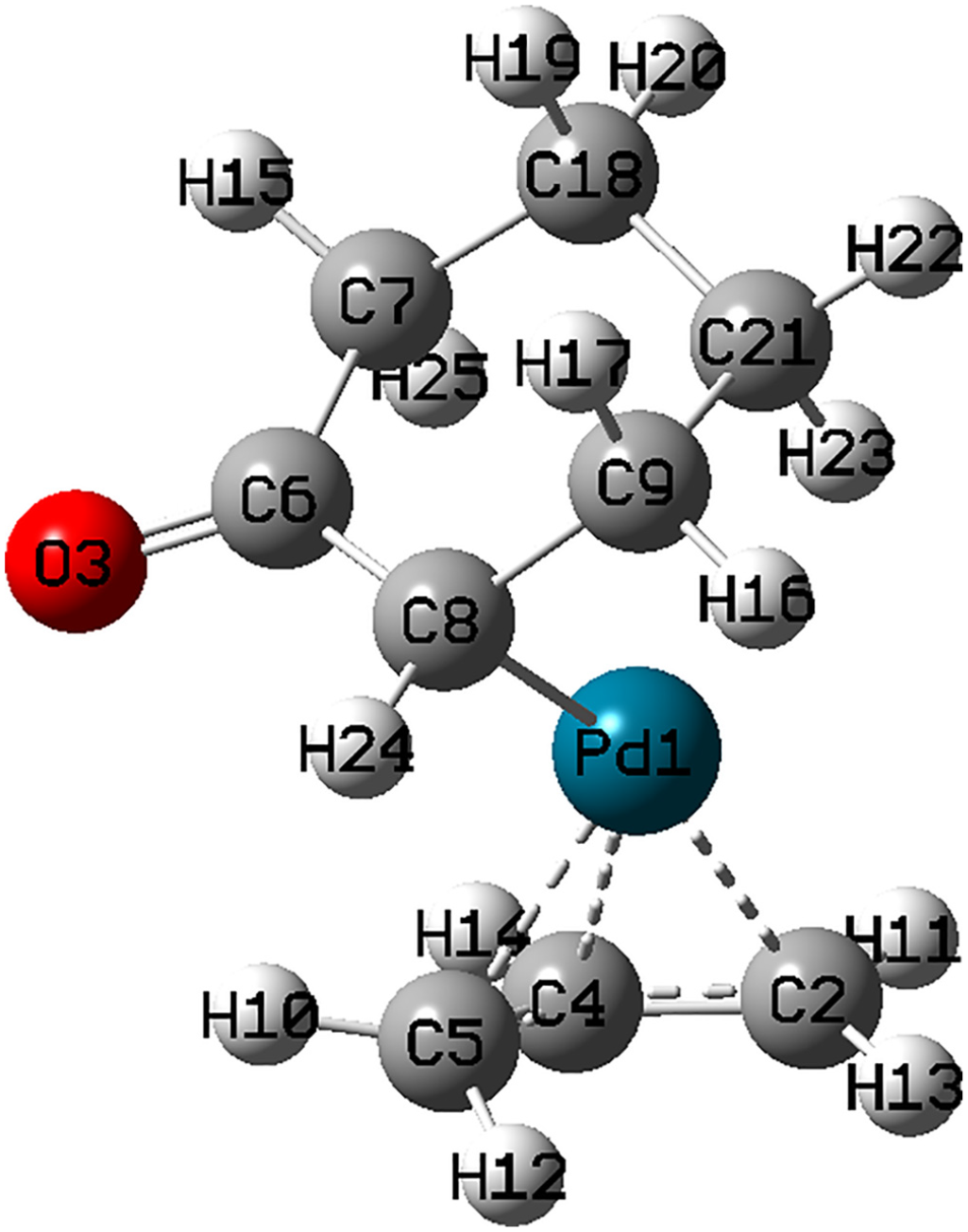
The optimized structure of complex
Selected bond lengths for complex

Complex
The calculated optimized structure of the transition state between complex
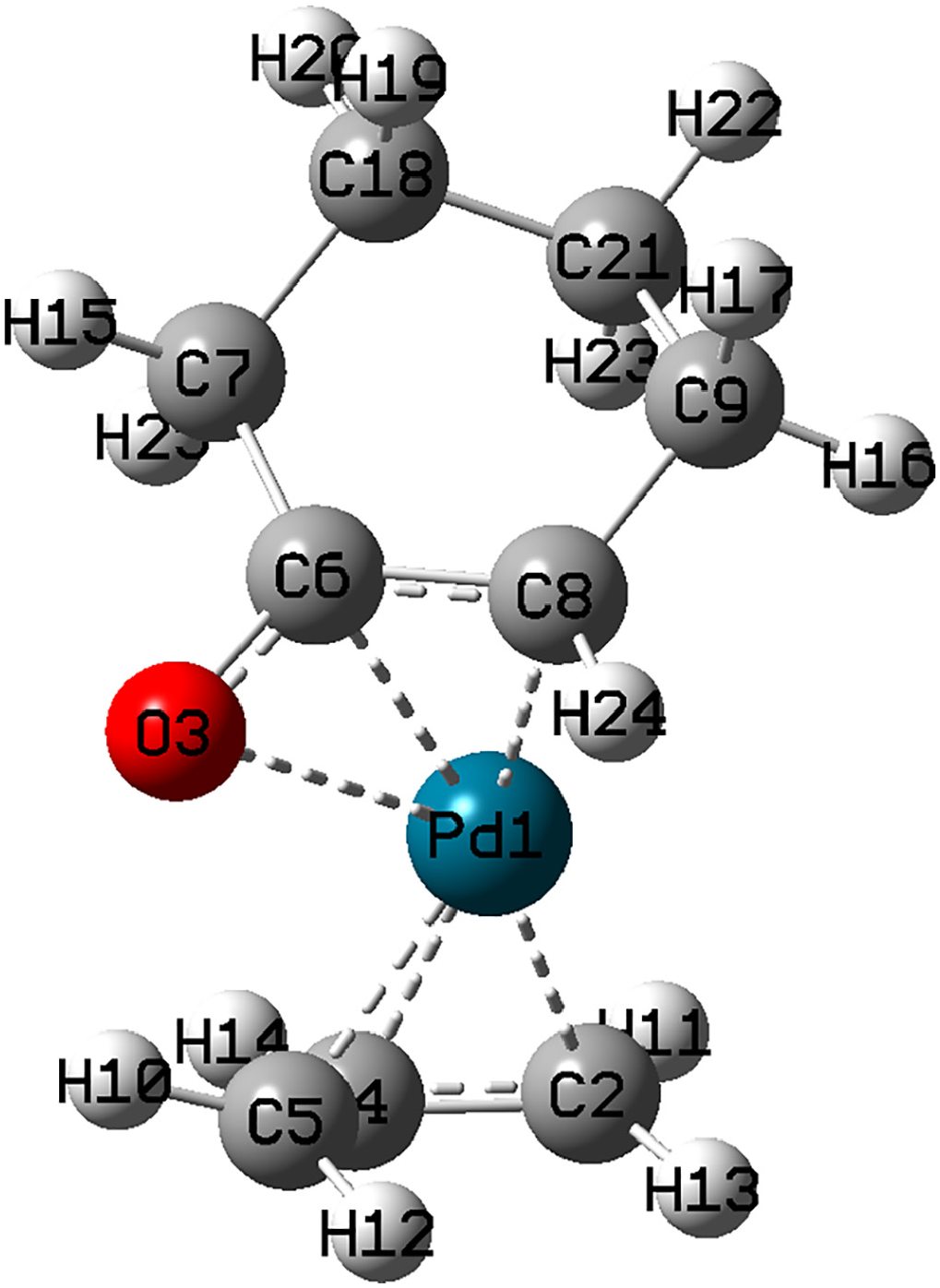
The optimized structure of
Selected bond lengths for

The results in Tables 2 and 3 indicate that the transition state,
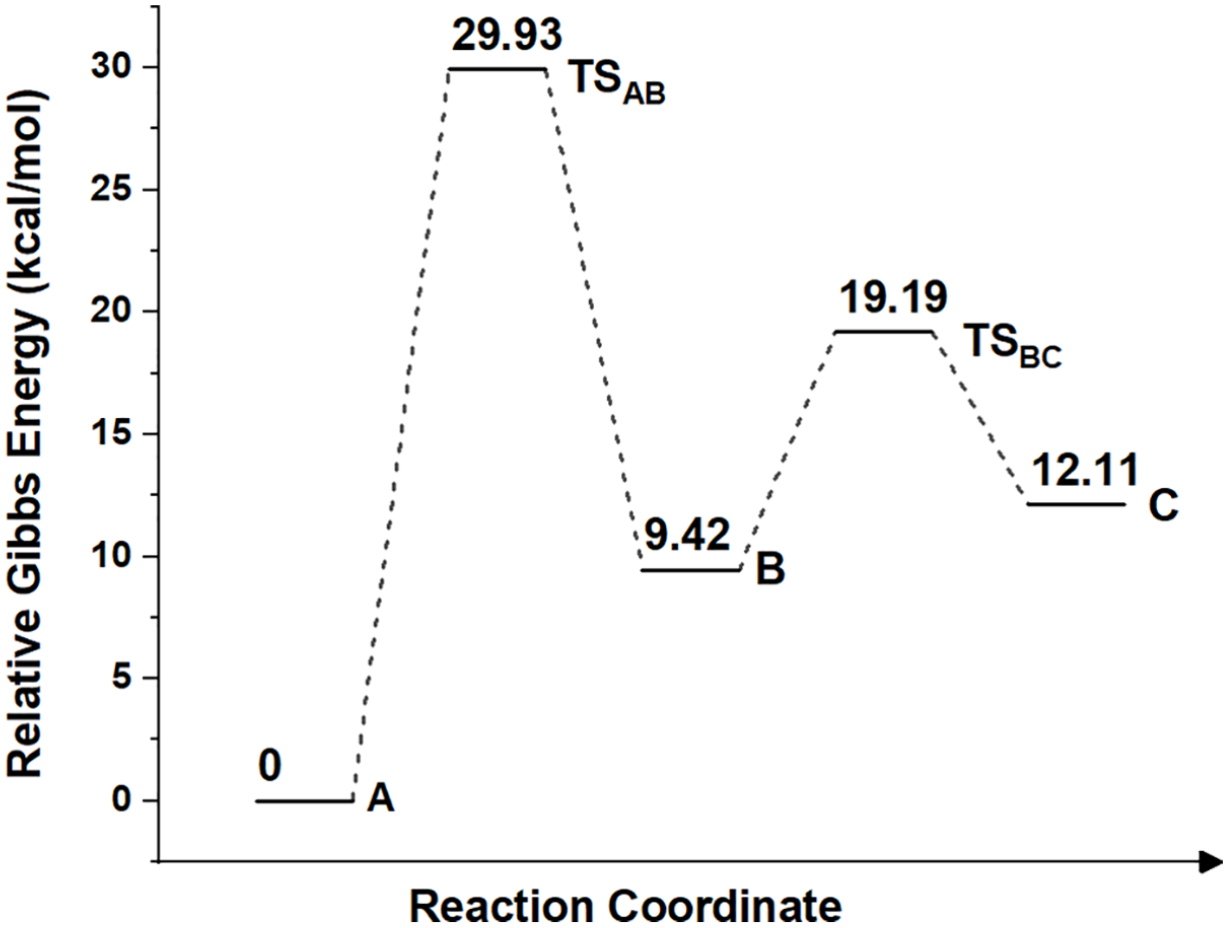
The relative Gibbs energy changes during the reaction in the gas phase.
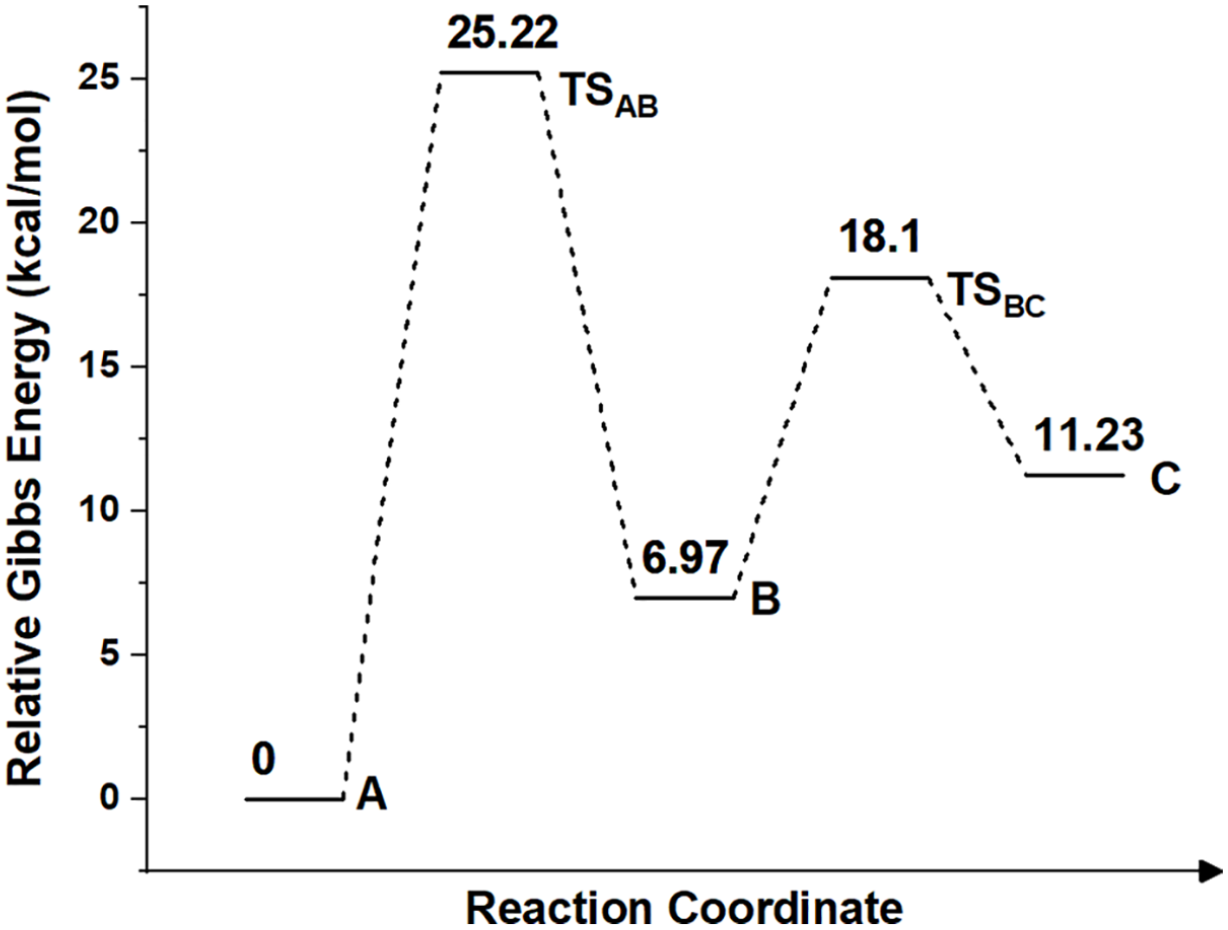
The relative Gibbs energy changes during the reaction in THF.
As the reaction continues, complex
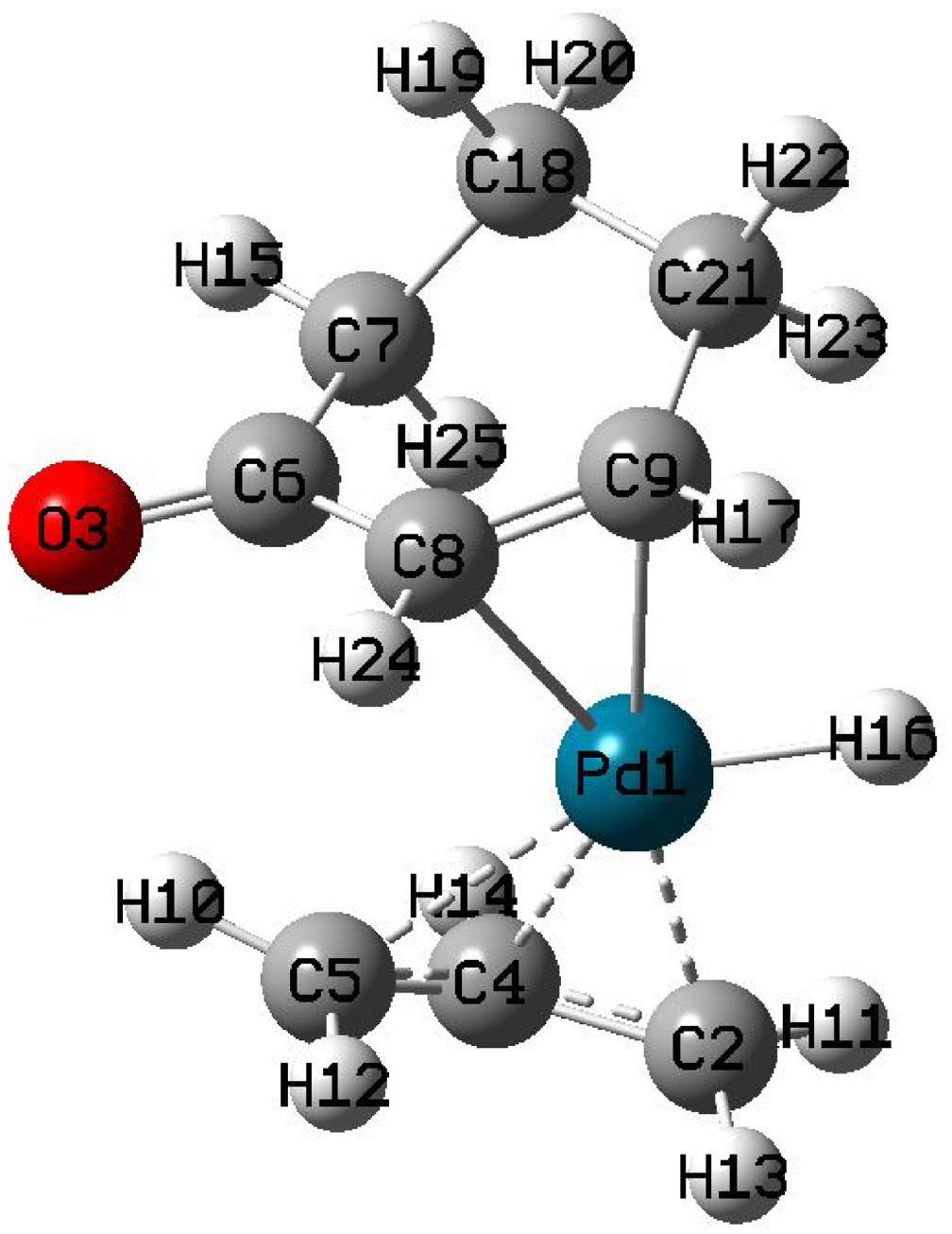
The optimized structure of complex
Selected bond lengths for complex

Complex
The optimized structure of the transition state between complexes
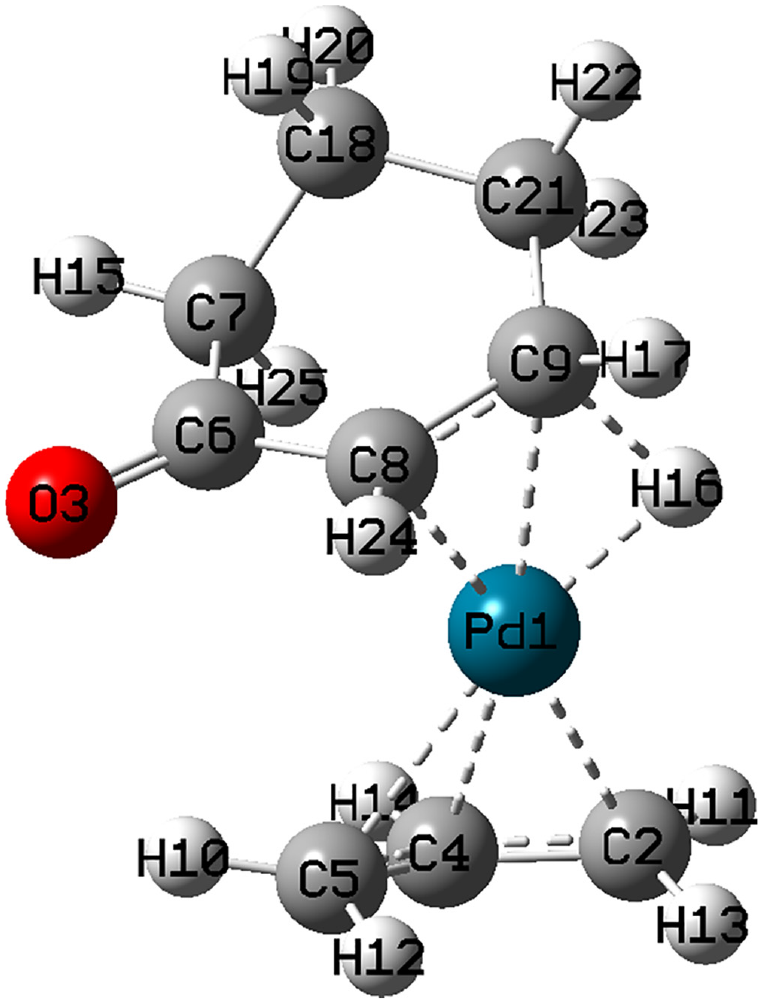
The optimized structure of
Selected bond lengths for

To confirm the proposed mechanism, the electrostatic potential map of the TSBC is presented in Figure 8.
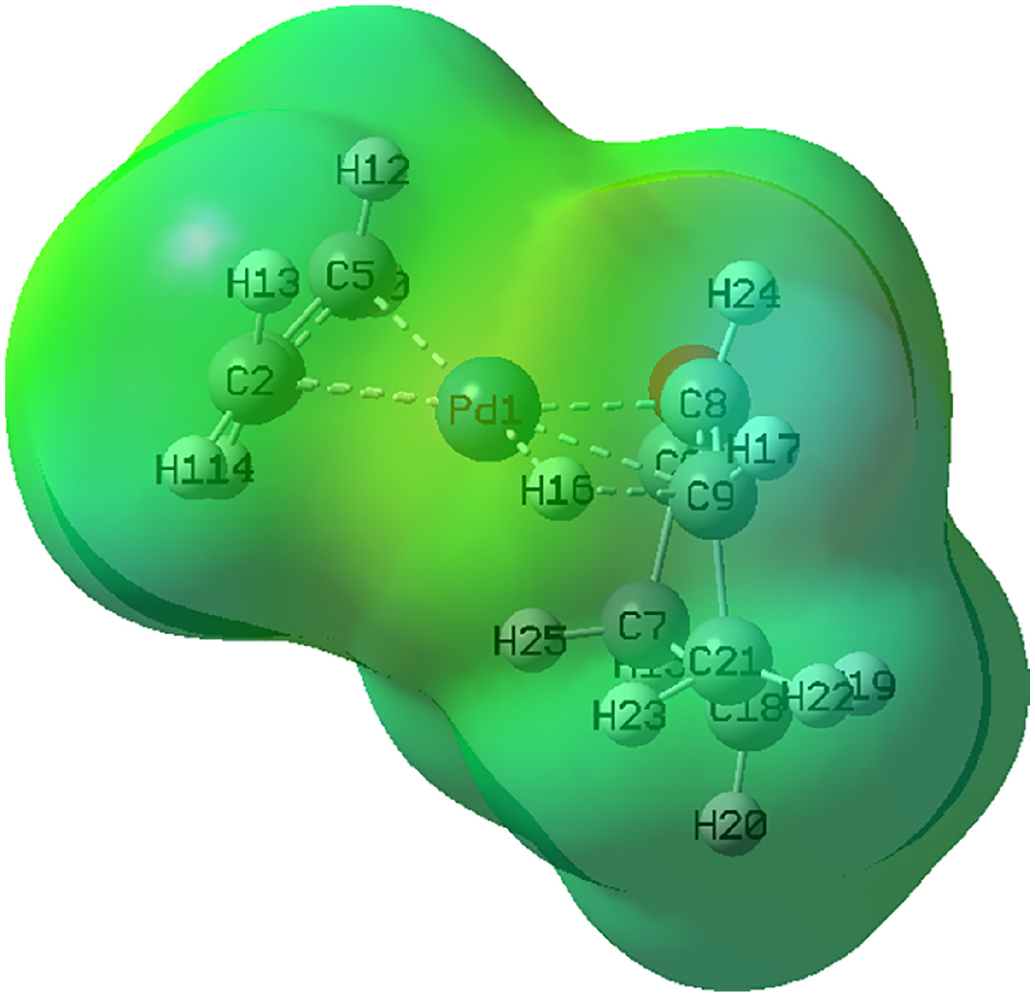
The electrostatic potential map of the
Here, it can be seen that H16 is more negative than H17, thus indicating the movement of H16 away from carbon and toward the Pd (i.e. a hydride transfer). Moreover, the highest occupied molecular orbital (HOMO) and the lowest unoccupied molecular orbital (LUMO) of complex
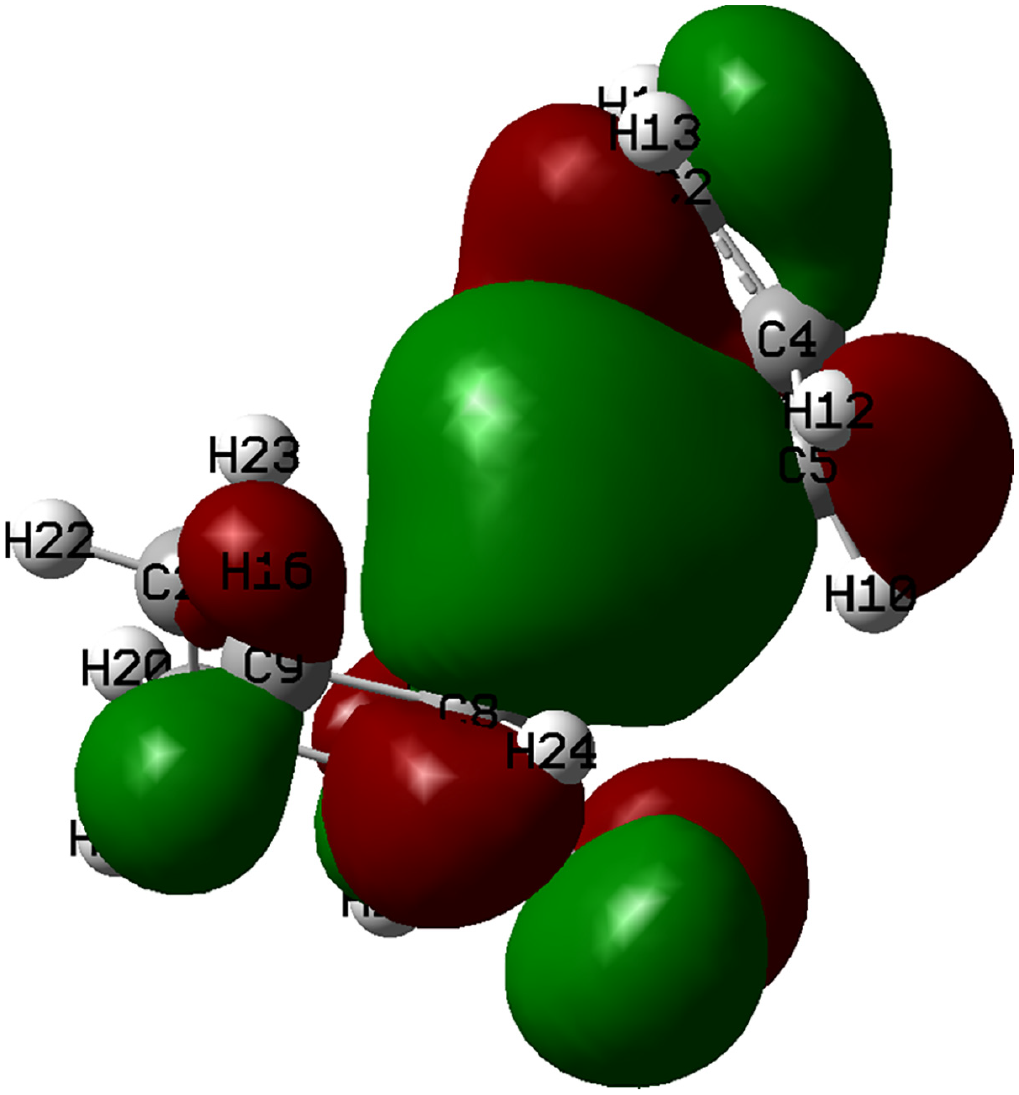
The HOMO orbital of complex
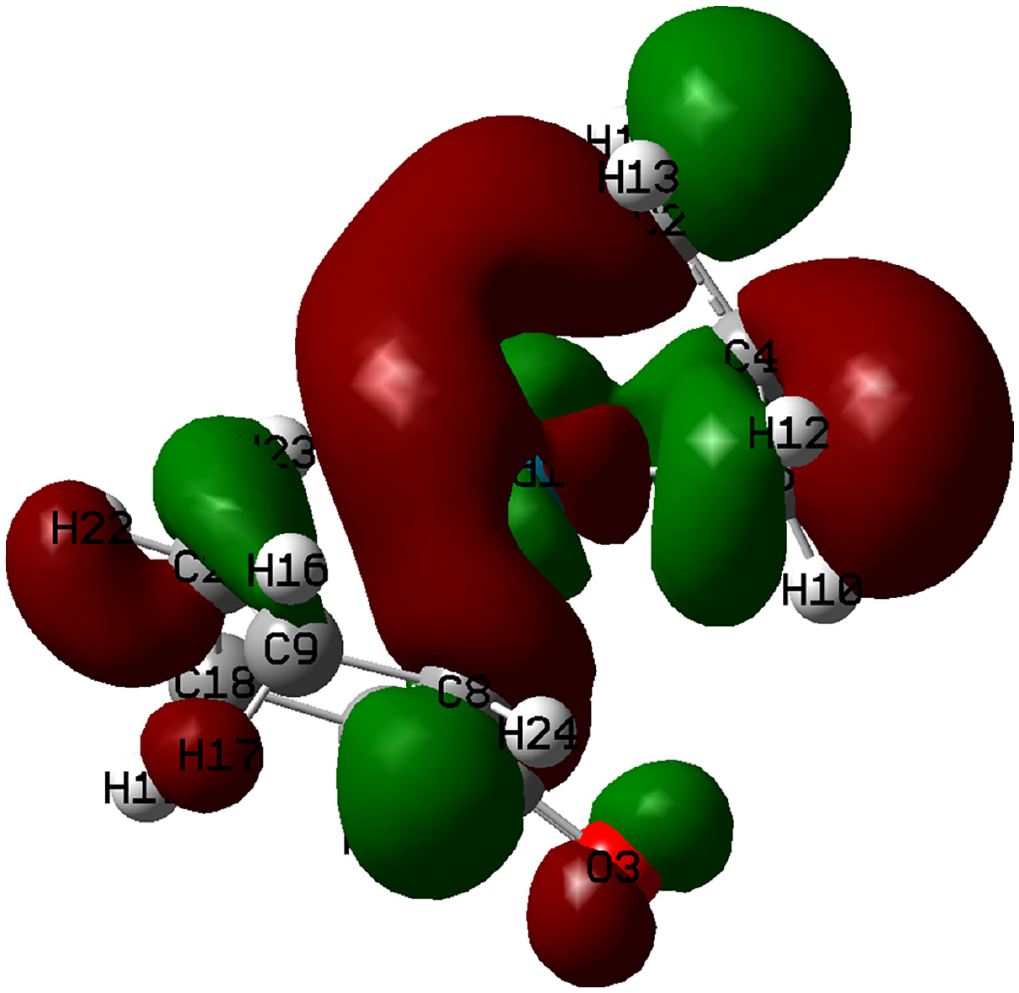
The LUMO orbital of complex
Conclusion
Herein, DFT calculations at the APFD/SDD level were performed in order to study the key steps in the mechanism of the α,β-dehydrogenation reaction of aldehydes and cyclic ketones catalyzed by an allylpalladium complex. The intermediates in this reaction (structures
Supplemental Material
sj-pdf-1-prk-10.1177_14686783211020600 – Supplemental material for A density functional theory study on the mechanism of the allylpalladium-catalyzed dehydrogenation of aldehydes and cyclic ketones
Supplemental material, sj-pdf-1-prk-10.1177_14686783211020600 for A density functional theory study on the mechanism of the allylpalladium-catalyzed dehydrogenation of aldehydes and cyclic ketones by Anan Haj Ichia Arisha in Progress in Reaction Kinetics and Mechanism
Footnotes
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.
Supplemental material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.