
Abstract
We have examined here the possibility of functionalization of the B12N12 cluster by methyl azide by means of a [2 + 3] cycloaddition reaction in analogy with the spontaneous functionalization of C20 fullerene using the same reaction. To achieve more reliable data, all possible interactions at different positions and orientations were considered by reaction channel study and potential energy surface calculations. Also, Born–Oppenheimer molecular dynamics simulations were used to find probable species which could emerge during the reactions.
Keywords
Introduction
The boron nitride (BN) fullerene-like clusters are one of the most important semiconducting materials which are expected to be widely applied in designing electronic components and nano-sized devices.1,2 Indeed, the future of nano-electronics could depend on developing such semiconductors. 3 Also, a wide range of BNs such as nanosheets with hexagonal rings (BN graphenes),4,5 cubic forms,6,7 wurtzite, 8 and fullerene-like clusters 9 have been introduced. There are ways of synthesizing BNs. For example, hexagonal BN was prepared by reacting boric acid or boron trioxide with urea or ammonia in a nitrogen atmosphere. 10 The cubic form of BN was synthesized by putting hexagonal BN under high pressure and temperature (much as synthetic diamond is formed from graphite). The direct conversion of hexagonal BN to the cubic form has been observed under similar conditions as for the direct graphite-to-diamond conversion. 11 Wurtzite-type BNs are produced by dynamic or static high-pressure shock procedures. Both cubic and wurtzite types are formed by compressing the graphite form of BN, but synthesizing of wurtzite occurs at much lower temperatures.12,13 Also the BN nanotube, which is structurally like the carbon nanotube, was predicted to exist in 1994 14 and then discovered experimentally in 1995. 15
In 2004, Oku et al. synthesized B12N12 clusters containing eight hexagonal and six tetragonal BN rings satisfying the isolated tetragonal rule, by the arc-melting approach from YB6 powder in an N2/Ar mixture atmosphere. 16 After successful synthesis of the B12N12 clusters, many investigations were performed on the properties and possible applications of these clusters, and the results revealed the exciting potential of these clusters.17–20
Due to these developments, in this work, we have tried to investigate the possibility of functionalization of the B12N12 cluster using its reaction with methyl azide. All possible interactions at different positions and orientations were probed by reaction channel approach calculations.21–24 Also, Born–Oppenheimer molecular dynamics (BOMD) simulations were applied to identify any possible species which could emerge during the reaction.25,26
Computational
A number of hypothetical geometries containing different orientations were developed as input files for each predicted species and were then optimized to give any reliable states. This resulted in finding a number of stable and metastable states for the reactants, products, and transition states (TSs). The Gaussian 03 package 27 was used to perform all the calculations, and density functional theory at the B3LYP/6-311G(d, p) level was used to optimize the possible structures.28–30 The TS structures were verified using the synchronous transit-guided quasi-Newton approach,31,32 and the frequencies of each optimized structure were extracted to calculate the thermodynamic parameters for each possible state. Intrinsic reaction coordinate (IRC) calculations were used to confirm the TS structures.33,34 To find the electrical charge of each atom in the reactants, products, and TSs, the natural bond orbital (NBO) analysis was used.35,36 The related partial bond order was then calculated through the Pauling relation (equation (1)) 37

where the bond order nx for a bond length rx is a function of a standard bond of length r0, whose bond order is defined as n0. The constant c determines, how steeply the bond orders change with bond distances.
The synchronicity relation38,39 was used to calculate the synchronicity40,41 of each considered pathway (equation (2))

where ∂Bi represents the relative variation of bond order index Bi at the TS.
The following formula (equation (3)) was applied to calculate the global electron density transfer (GEDT) 42

where qA is the net Mulliken charge and the sum covers all the atoms of the dipolar species. The simulations were performed using the BOMD method, which is widely used for similar calculations. 43
Results and discussion
Following reports about the rare possibility of existing stepwise routes and emerging intermediates during [2 + 3] cycloaddition reactions, 44 some structures referring to probable intermediates for the stepwise routes were designed and optimized to give the true structures. For reactants, TSs, and products, several geometries have been developed as input files and then optimized to provide the stable or metastable states. Some reliable structures have been detected for the reactants, a number of products, and the TSs of the concerted routes (Figure 1).
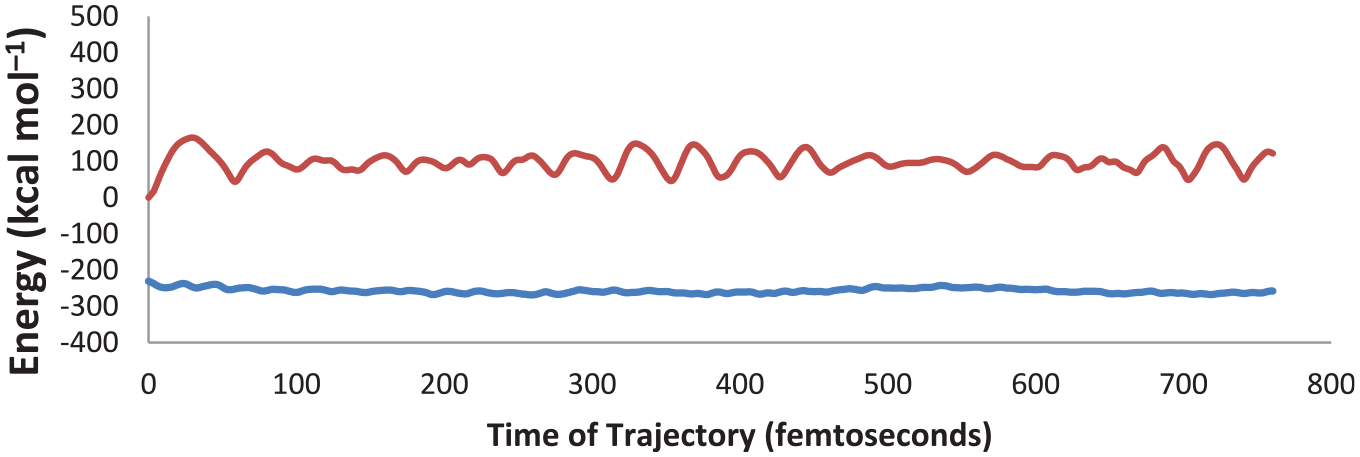
The reason for the formation of TS (Con-E) instead of TS (Con-F) may be due to opening of the comprised four-membered ring in TS (Con-E) during production of product E (while in TS (Con-F), the two neighboring rings are the usual six-membered ones). Furthermore, in the isolated cluster, the nitrogen and the boron atoms have partial negative and positive charges, respectively. Since, in the isolated methyl azide, the terminal nitrogen has a partial negative charge, this nitrogen could not attach to the nitrogen atoms of the cage. Thus, the TSs for the routes A′ and B′ would not form.
However, attempts to find a reliable intermediate for the stepwise route failed. Thus, the concerted reaction pathway was chosen for further consideration as a result of the dominance of the concerted routes (Scheme 1). Also, it should be noted that all calculations were performed at the B3LYP/6-311G(d, p) theoretical level (Figure 1).
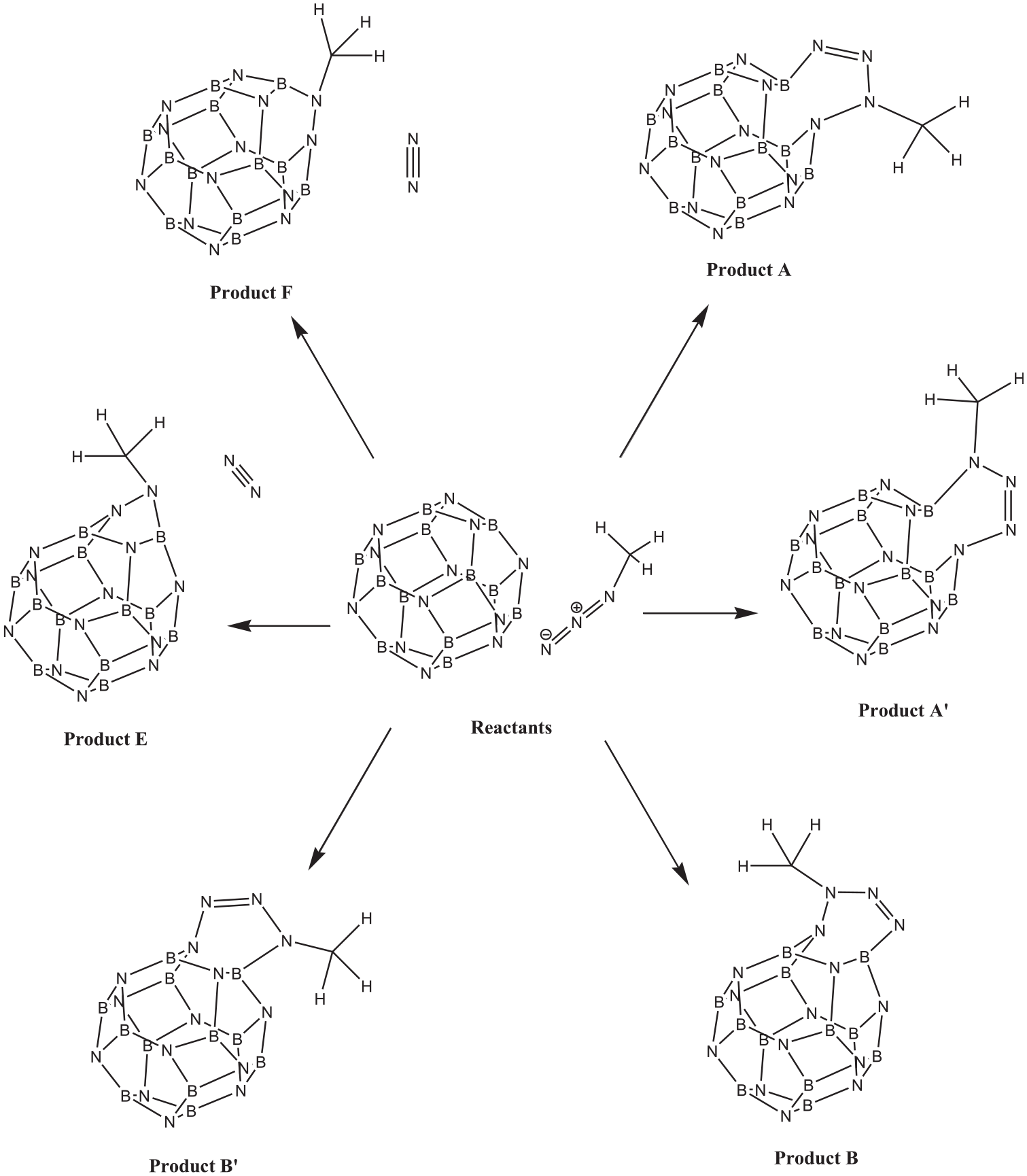
Proposed reaction pathways for interactions between B12N12 cluster and methyl azide.
During the optimization of the reactants (a number of input files with different orientations), two geometries were found. One of those two had a 2.88 Å distance between a hydrogen atom of methyl azide and a nitrogen atom (N21) of the cluster (“Reactants” in Figure 2), and the other one had a 2.54 Å distance between the terminal nitrogen atom of the azide (N27) and a boron atom of the cluster (B19) (“Reactants complex (RC)” in Figure 2). It seems that before the reaction, in the first step, RC is formed from the free reactants, and then, the TSs form. For probable cycloadditions, to different positions, containing sites A and B were considered. Position A is the B–N bond between the hexagon–hexagon rings, and the position B is the B–N bond between the square–hexagon rings. TSs B and E refer to cycloadditions at position B (Figure 2).
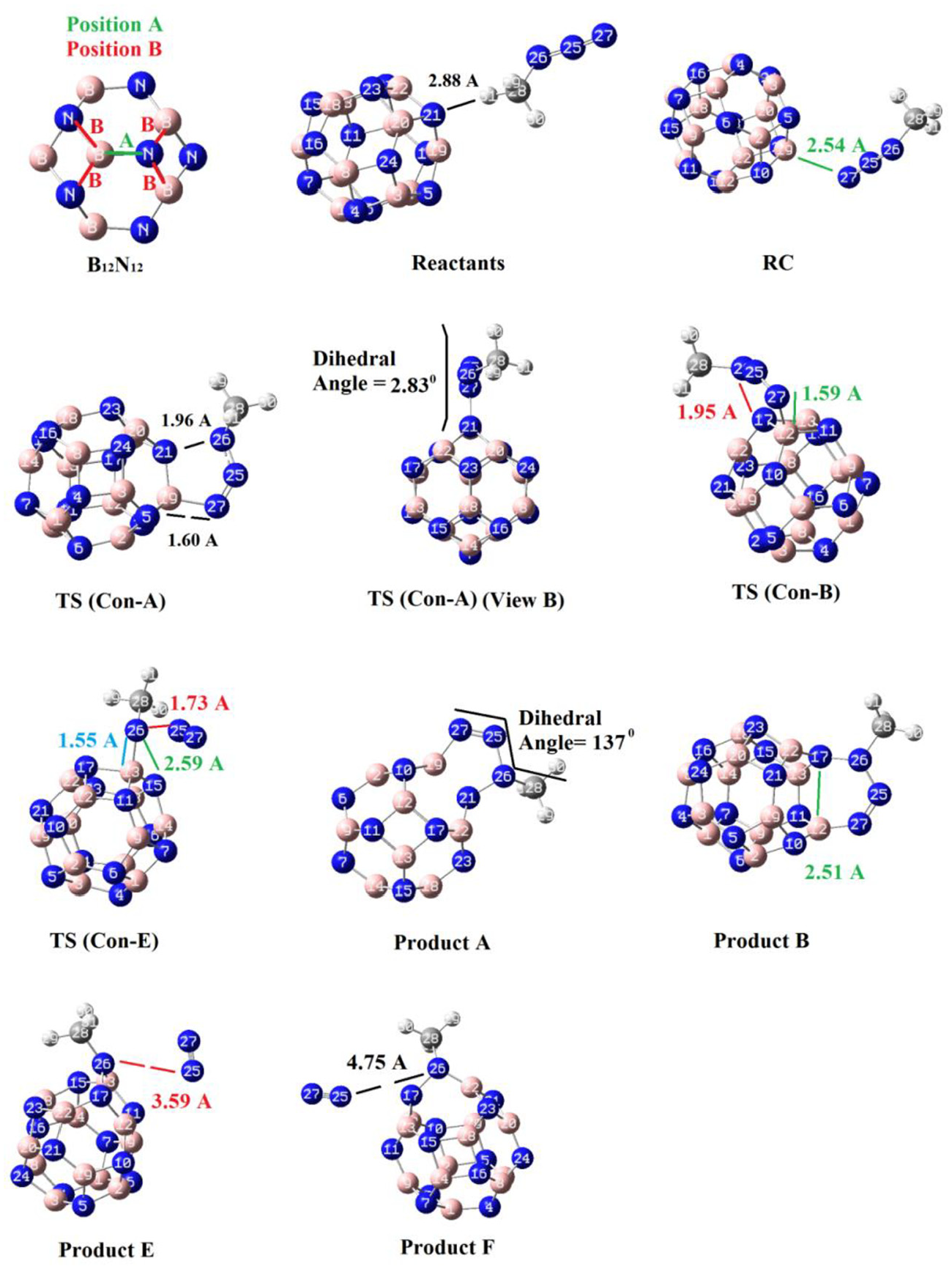
Optimized structures of the detected species which emerge during the interactions (calculated at the B3LYP/6-311G(d, p) theoretical level).
Some previous reports revealed that [2 + 3] cycloaddition reactions between the 1,3-dipoles and C20 fullerene (or its derivatives) as dipolarophile proceed in a very fast and spontaneous manner.45–50 However, in this project, it was observed that the [2 + 3] cycloaddition between the B12N12 fullerene-like cluster (as dipolarophile) and methyl azide (as dipole) has a ΔG# of 29.38 kcal mol−1 (on position A) and 31.95 kcal mol−1 (on position B). Thus, [2 + 3] cycloaddition of azide to the B12N12 cluster occurs very slowly or is nearly impossible. Rather, instead, a [1 + 2] cycloaddition could occur at position B with a ΔG# of 14.65 kcal mol−1 (Figure 2 and Table 1). On the other hand, TS (Con-A) and TS (Con-B) refer to [2 + 3] cycloadditions at positions A and B, respectively, while TS (Con-E) refers to [1 + 2] cycloaddition at position B (leaving an N2 molecule). Our attempts to find TSs for [2 + 3] cycloadditions at sites A or B with orientations opposed to TS (Con-A) or TS (Con-B) (when the terminal nitrogen of azide is attached to a nitrogen of the cluster) failed (TS (Con-A′) and TS (Con-B′) in Scheme 1, Figure 2). The transfer of electronic charge from the terminal nitrogen of the azide to the boron atom of the cluster, which is discussed in the following section, might be a reasonable explanation for this. Attempts to find a TS for [1 + 2] cycloaddition on position A (giving product F) were unsuccessful. Also, the entropies of reaction for routes Con-A (−29.64 cal mol−1 K−1) and Con-B (−28.14 cal mol−1 K−1) are highly negative. Thus, these reactions are very unfavorable compared with the reaction for route Con-E (entropy of reaction −6.04 cal mol−1 K−1).
Key thermodynamic and kinetic parameters for the possible reactions between the B12N12 cluster and methyl azide.
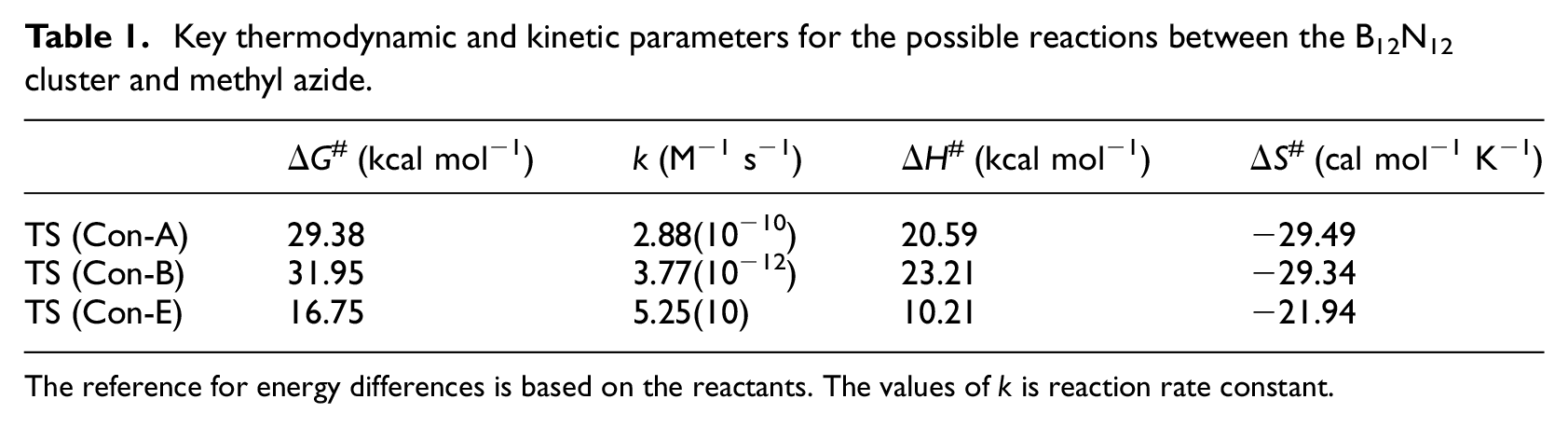
The reference for energy differences is based on the reactants. The values of k is reaction rate constant.
Table 2 shows that the concerted mechanism for route Con-A is initiated by the two key bond formations of B19–N27 and N21–N26 (1.60 and 1.96 Å, respectively). The obtained partial bond orders (which were calculated by the Pauling relation) 37 for those bonds were 0.766 and 0.188, respectively. The obtained partial bond orders indicate that 76.6% and 18.8% of bonds B19–N27 and N21–N26 are reached in the TS, respectively. The mechanism for route Con-B is initiated by the two bond formations of B12–N27 (1.59 Å) and N17–N26 (1.95 Å), which have partial bond orders of 0.792 and 0.195, respectively. Therefore, 79.2% and 19.5% of bonds B12–N27 and N17–N26 are reached in the TS, respectively. The synchronicity values of route Con-A (0.802) and route B (0.809) were almost the same, and those two are asynchronous.
Key parameters for critical structures of [2 + 3] cycloaddition reactions involving B12N12 cluster and methyl azide in gas phase.
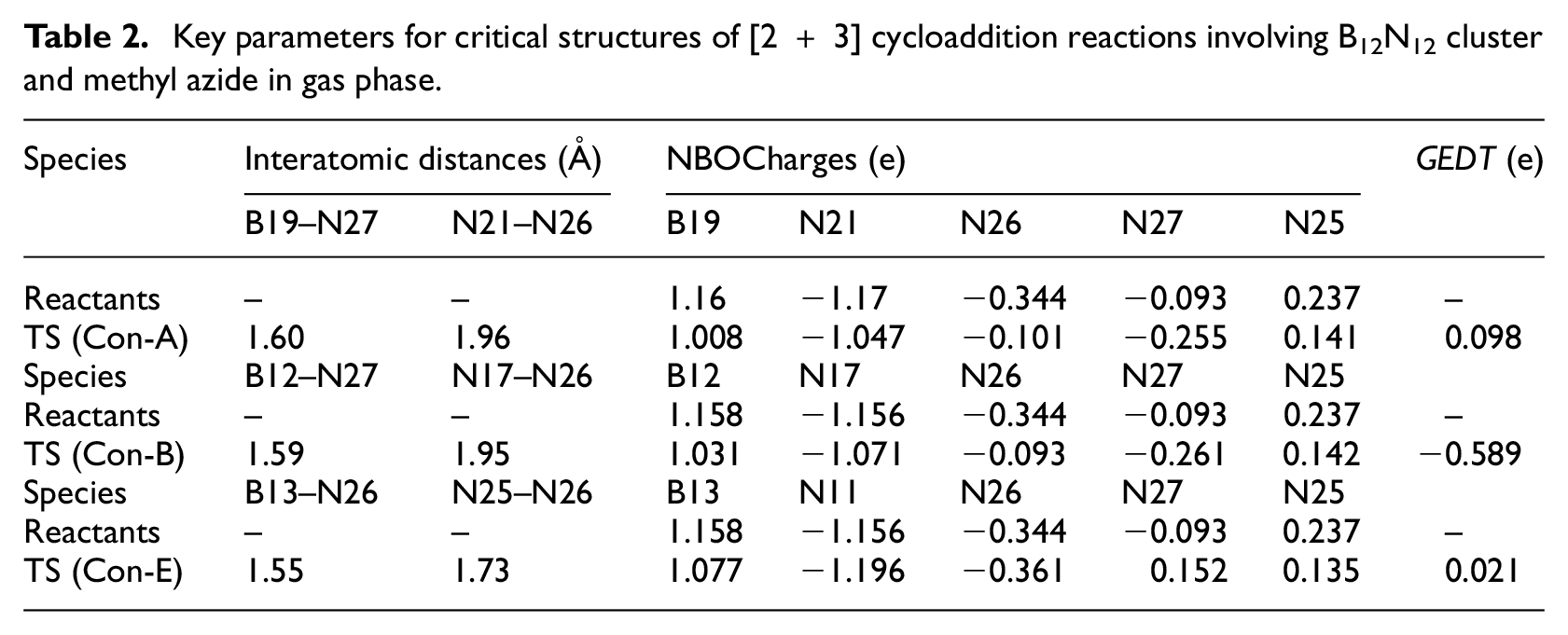
Route Con-E, which results in production of product E via a [1 + 2] cycloaddition, is begun by formation of B13–N26 (1.55 Å) and N15–N26 (2.59 Å) and breaking of N25–N26 (1.73 Å) bonds. The partial bond orders for these bonds were 0.904, 0.023, and 0.407. Thus, the TS contains 90.4%, 2.3%, and 40.7% of bonds B13–N26, N15–N26, and N25–N26, respectively. The calculated synchronicity value of the route (0.711) shows that this reaction route is highly asynchronous.
The orbital imaging calculations showed that in the reactants, the highest occupied molecular orbital (HOMO) is localized on the dipole (methyl azide), while the lowest unoccupied molecular orbital (LUMO) is mostly localized on the cluster. Therefore, it could be concluded that the electron density moves from the HOMO of methyl azide to the LUMO of the cluster. That is, the electrons of the HOMO of methyl azide attach to the LUMO of the cluster (Figure 3). The localization of HOMO orbitals on the azide part is also observed for the TSs. Again, the results of the GEDT (+0.021 e, see Table 2) and the energy surfaces of the HOMO and LUMO confirm the predictions. The HOMO and LUMO energies of the cluster are −0.280 and −0.057 eV, and the HOMO and LUMO of the azide are −0.260 and −0.031 eV, respectively. The HOMO–LUMO gap is lower when the HOMO is on the azide and the LUMO is on the cluster; thus, frontier molecular orbital calculations confirm that the electron charge transfers from the azide to the cluster (Figure 3).
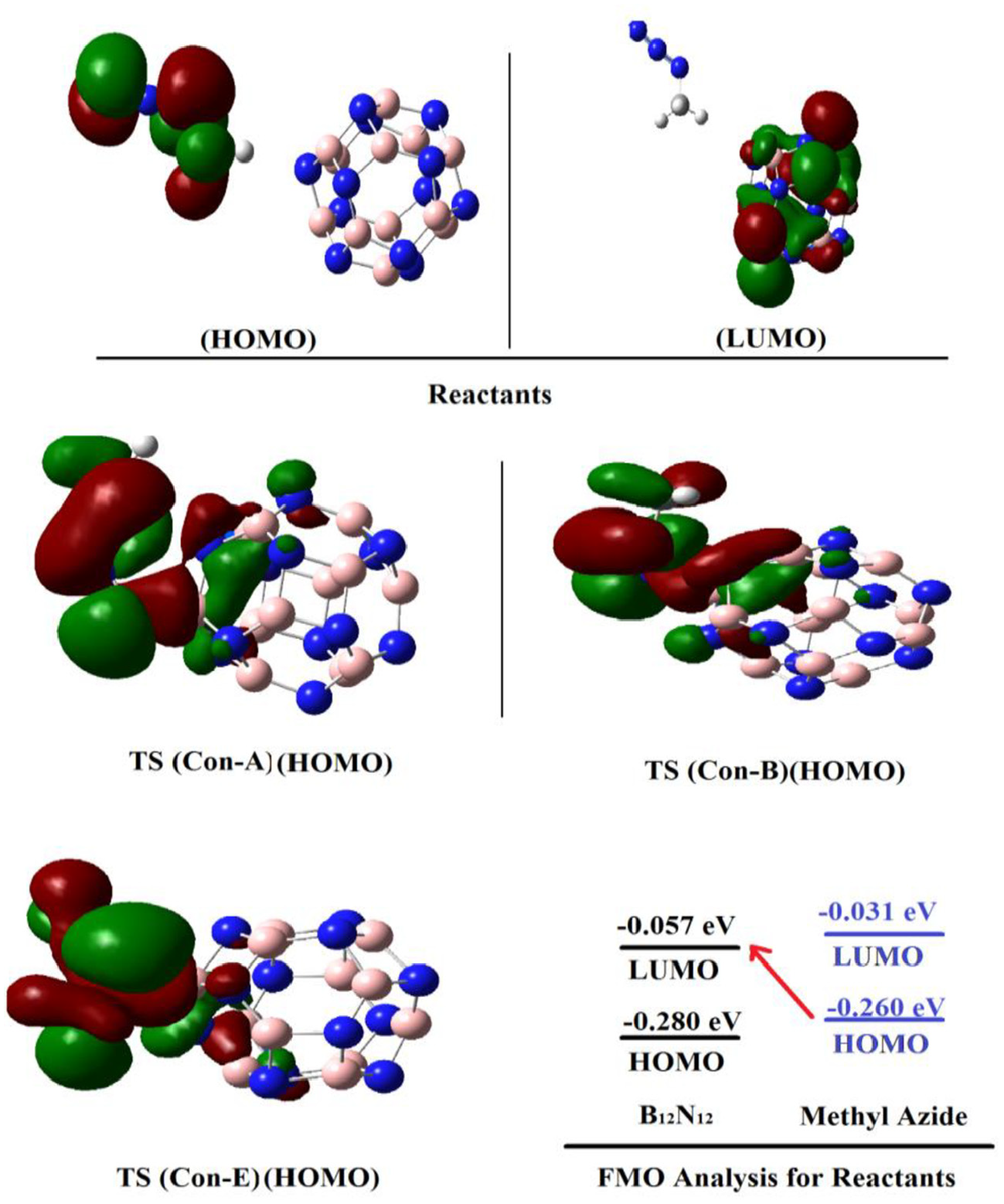
Shapes of the key molecular orbitals (HOMO and LUMO) for selected species.
Conclusion
Previous reports revealed that [2 + 3] cycloaddition reactions between 1,3-dipoles and C20 fullerene proceed by very fast and spontaneous routes. However, in the present work, the results show that the [2 + 3] cycloaddition between the B12N12 fullerene-like cluster and methyl azide (as dipole) has a ΔG# of 29.38 kcal mol−1 (on position A) and 31.95 kcal mol−1 (on position B). Therefore, the [2 + 3] cycloaddition of azide on the B12N12 cluster occurs very slowly (i.e. is virtually impossible). However, a [1 + 2] cycloaddition could occur at position B with a ΔG# of 16.75 kcal mol−1. The calculations also showed that the [1 + 2] cycloaddition reaction is highly asynchronous, and the orbital imaging calculations showed that in the reactants, the HOMO is localized on the dipole (methyl azide), while the LUMO is mostly localized on the body of the cluster. Thus, the electron density flows from the HOMO of methyl azide to the LUMO of the cluster. The calculated synchronicity value of route Con-E (0.711) shows that this reaction route is highly asynchronous. Finally, as opposed to our prior predictions, a [1 + 2] cycloaddition (with a leaving N2 molecule) is favorable instead of the expected [2 + 3] cycloaddition between the B12N12 cluster and methyl azide.
Footnotes
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.