
Abstract
The tautomeric equilibrium of benzimidazolone and benzimidazolthione have been studied by the density functional theory method using the CAM-B3LYP functional together with the 6-311G(d,p) basis set. Two separate mechanisms have been investigated: a direct intramolecular transfer using the polarizable continuum model and an indirect proton transfer assisted by a molecule of solvent (C6H12, H2O, CH3OH, and H2O2). In both cases, the results obtained indicate that ketone and thione are the most stable forms. However, the enhanced height of the activation barrier for the four-center mechanisms describing the tautomerism reaction as a direct intramolecular transfer implicates a relatively disadvantaged process. The participation of a polar protic solvent molecule allows the lowering of the activation energy barrier. Potential energy profiles of keto-enol and thio-enol tautomerism assisted by methanol and water are very different. The former one describes a concerted mechanism but the latter does not because it is associated with asynchronous processes that take place during the thio-enol tautomerism.
Keywords
Introduction
Proton transfers are one of the simplest and fundamental known reactions, being crucial for several biological and chemical processes.1–5 They also play an important role in the interpretation of several reaction mechanisms. 6 These processes occur often in aqueous solutions and are able to effect the transfer of one or more protons every time. Multiple proton transfers appear when several hydrogen atoms are allowed to move from one atomic center to another simultaneously or by steps. 7 This type of reaction occurs in aqueous complexes establishing hydrogen bonds and in several enzymes. Furthermore, these reactions are very sensitive to any changes in their environment.
The migration of a hydrogen atom creates different structures, generating a tautomeric equilibrium that plays a very important role in many organic and biochemical reactions. It intervenes in the biological and chemical behaviors of several compounds.8–14 The composition of the tautomeric mixture is often difficult to determine. 15 The tautomeric equilibria encountered are of the type: keto–enol; enamine–imine; acinitro-nitro, nitroso–oxime; and the transformations of the amide–iminole, 16 thione, and thiol.17–19
In structural chemistry, knowledge of the relative stability of the tautomeric species and their conversion from one to another seems to be very important. 20 The determination of their geometrical and electronic structures, as well as the stability of the tautomeric forms, leads to an understanding of their biological activities and pharmacological properties. 21
The tautomerism of heterocycles has been widely studied for its importance in biology and for its high dependence on solvents. 22 It has been shown that solute–solvent interactions can determine the relative stability of the tautomeric forms. 23 Numerous studies have demonstrated that tautomeric equilibria can be influenced by the nature of the solvent.24–27
Benzimidazolones are heterocyclic compounds containing two nitrogen atoms and carbonyl groups (Figure 1). This particularity of their structures confers a considerable place to these compounds in different areas. Thus, they are used as anti-HIV,28,29 antitumor, 30 anti-ulcer 31 and antihistamine drugs. 32 They are also associated with several pharmacological properties.33–36 Despite the existence of many methods for the synthesis of benzimidazolones, there is a real need for new simple procedures that support many types of structural diversity. 37 The synthesis of new substituted benzimidazolones, such as benzimidazolone and benzimidazole-2-thione, has recently been realized. 38
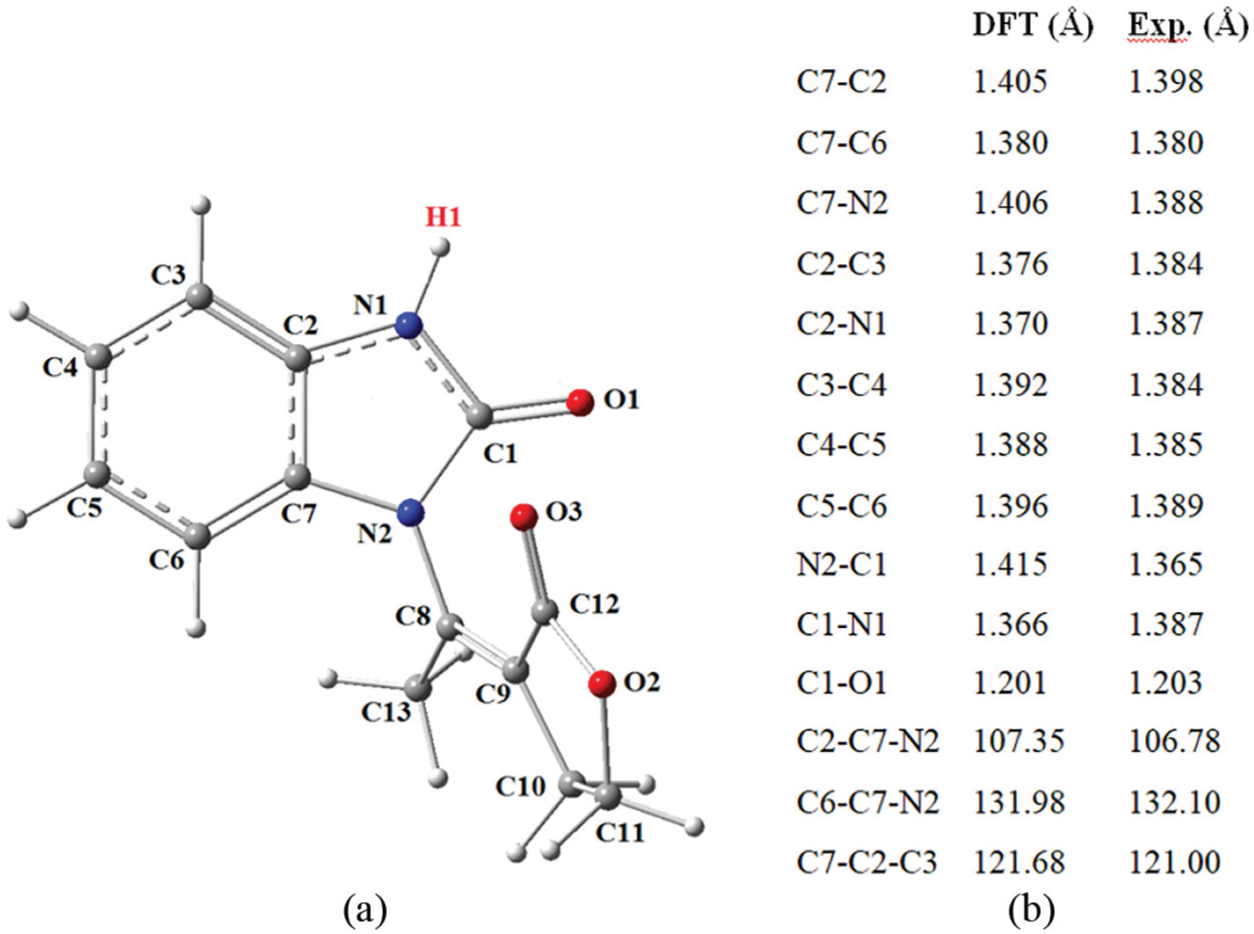
(a) Optimized geometry of benzimidazolone (b) Geometric parameters of benzimidazolone optimized by DFT method/Cam-B3LYP and the X-ray data for an analogous compound. 18
Benzimidazole and benzoxazole-2-thione derivatives act as inhibitors of bacterial hyaluronan lyase. 39 Indeed, depending on the position of the hydrogen atom on nitrogen or on sulfur, there is a competition during the alkylation reactions of RX (R is an organic radical and X is oxygen or sulfur) in basic medium between the substitution of the radical R on the nitrogen and its addition to the sulfur. 18 Controlling this competition requires an understanding of the tautomeric process thione–thiol. However, it is well established that the antibacterial activity of substituted benzimidazolones against several strains of bacteria is very sensitive to the nature and position of the ligand. 40
Our work focuses on many problems, especially the effect of oxygen substitution by sulfur on proton transfer tautomeric equilibrium. The mechanisms governing these exchanges of protons are complex and still poorly understood despite the existence of several kinetic41–43 or theoretical studies.43–46 In this study, we aimed to make a thorough exploration of the proton transfer mechanism intervening on these new molecules. First, we focused on the direct transfer of a proton using different theoretical calculation approaches. Second, we dealt with the assistance effect of polar protic solvents on this tautomeric equilibrium. Finally, we were interested in the evaluation of the effect of substitution of oxygen by sulfur.
Computational details
Molecular geometries of reagents, transition states (TSs) and products have been optimized using the Cam-B3LYP 47 functional together with the 6-311G(d,p) basis set. 48 For each system, we have conducted a full optimization of the geometry. The different calculations of this work were performed using the 9th version of the GAUSSIAN program. 49 Frequency calculations were carried out for all the optimized geometries in order to differentiate the minima and the transition states TSs. The calculated harmonic vibrational wavenumbers were scaled with 0.9665 for CAM-B3LYP/6-311G(d,p) level. 50 We have also performed calculations in three different solvents (ε = 2.3, benzene C6H6); (ε = 32.63, methanol CH3OH); and (ε = 78.39, water H2O) using the polarizable continuum model (PCM).51,52 The thermodynamic quantities have been calculated at 298.15 K for all the optimized geometries.
Results and discussion
Intramolecular tautomerism process
In this part, we studied the intramolecular tautomerism reaction of benzimidazolone and benzimidazolthione (denoted by the letters O and S, respectively). G, W, and M designate compounds in the gaseous state or assisted by a molecule of water, methanol, or Hydrogen Peroxide (H2O2), respectively. Before we started the study of the reaction mechanism itself, we compared our model structure of benzimidazolone to the experimental structure of a similar compound. 18 Calculated geometric parameters are gathered in Figure 1. The analysis of these values showed a sufficient agreement with experiment which validates the choice of calculation method.
The geometric parameters of the reagents, TSs, and products are shown in Figure 2. The data for the thermodynamic quantities in the gas phase and in various solvents are summarized in Table 1. The calculated Gibbs energy of the reaction in the gas phase is about 14.80 kcal mol−1.
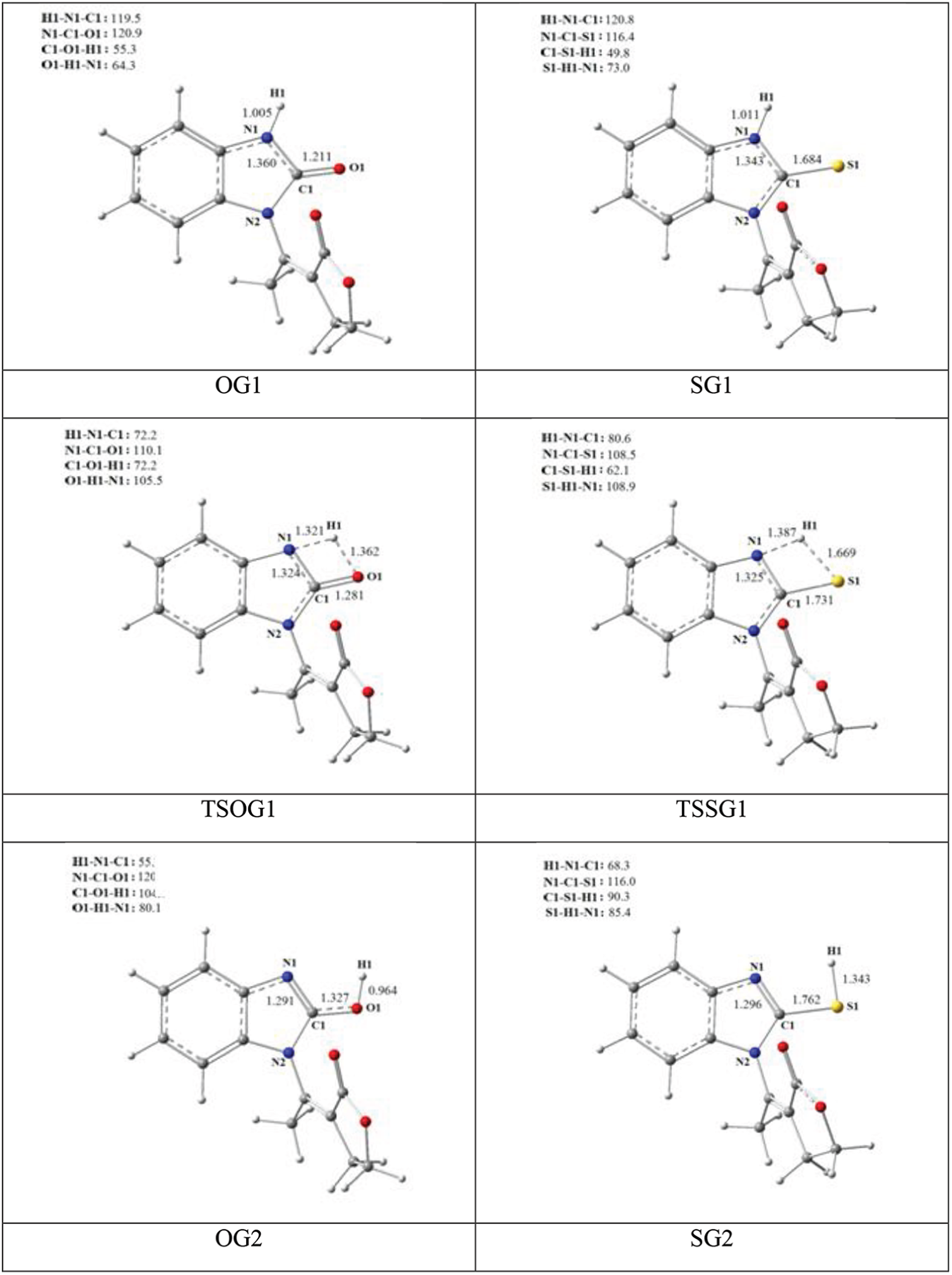
Structural parameters of reagent, transition state, and product of tautomerism of benzimidazolone and benzimidazolthione.
Thermodynamic and kinetic parameters in kcal mol−1 of the tautomeric reaction of benzimidazolone and benzimidazolthione in various solvents.
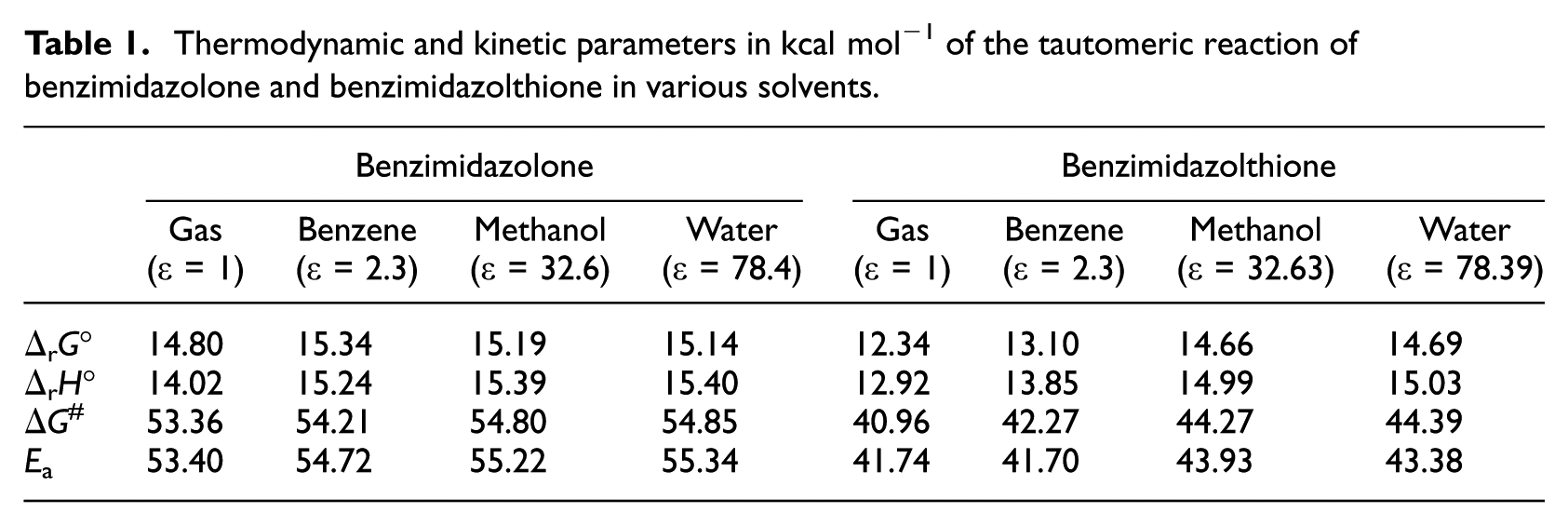
We determined a transition state, denoted TSOG1, which intervenes in the process described by direct transfer of the hydrogen atom bonded to the nitrogen atom N1, to the oxygen atom O1. Intrinsic Reaction Coordinates (IRC) are generally used to ensure that the TS does indeed connect the reactant and product. Our calculations verify this assertion, as is shown in Figure 3(a). The activation barrier of TSOG1 is 53.40 kcal mol−1. This value can be considered as so high as to be inaccessible. These results are in agreement with those reported in previous studies on other molecules. 46 When the benzimidazolone molecule is replaced by benzimidazolthione, the result obtained shows clearly that the thione form is more stable than the thiol one. As in the case of benzimidazolone, such large variation makes proton transfer difficult, showing a high barrier (41.74 kcal mol−1) to tautomerism. It is lower by about 11.66 kcal mol−1 than the benzimidazolone tautomerism activation barrier described previously in the gas state. Such a high barrier was also observed in other theoretical studies on similar molecules.44,45
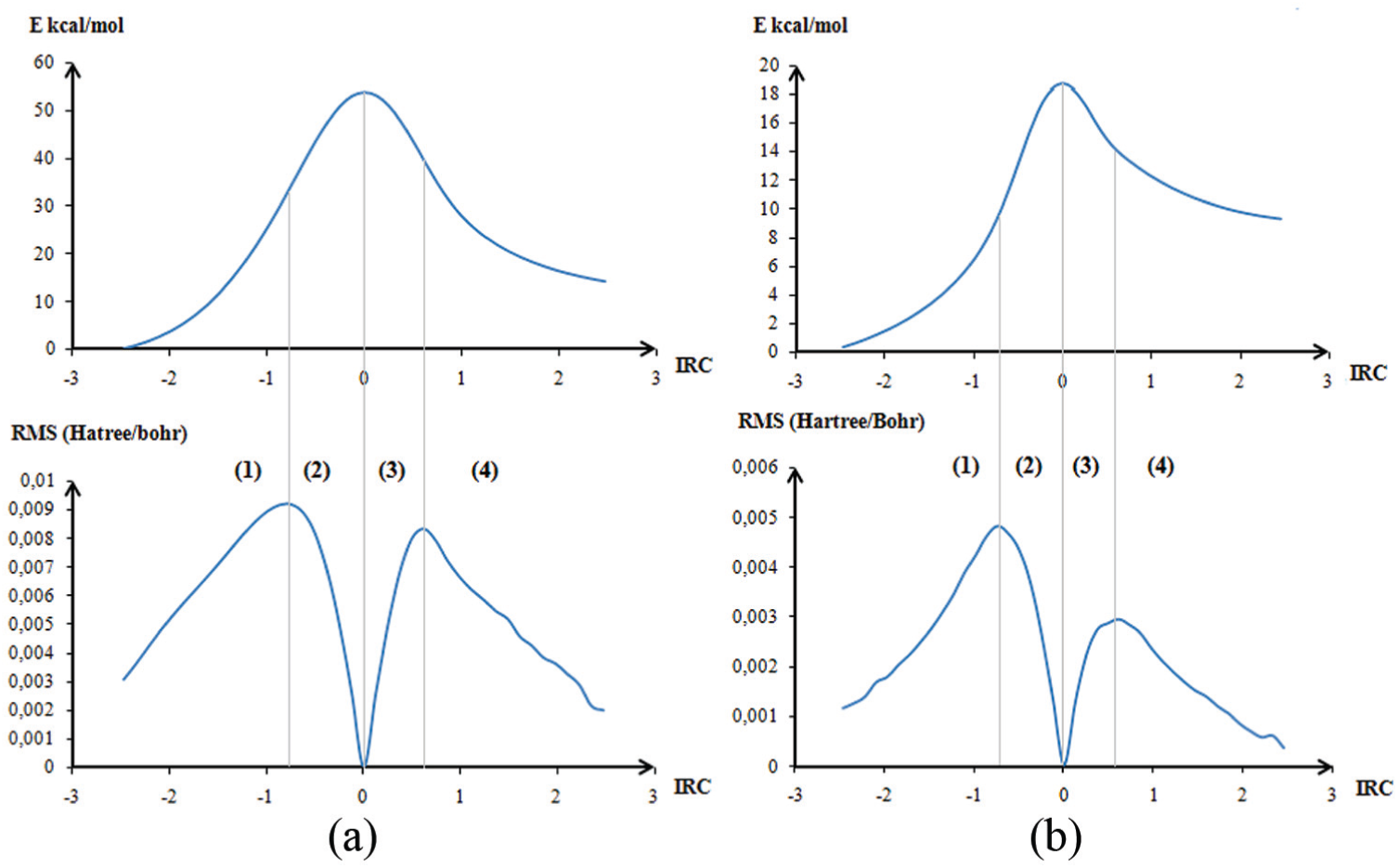
Energy profile and its first derivative of tautomeric benzimidazolone: (a) in gas phase and (b) assisted by a molecule of water. (1), (2), (3) and (4) are the different parts of the curve.
Very often, the difference between a value calculated theoretically and the experiment is interpreted by the fact that the first concerns the gas state and the second has been obtained in the liquid state. To test the validity of this argument, we have repeated the calculations in the liquid state by using the model of implicit solvation, i.e., the PCM of Tomasi et al. 51 The results are summarized in Table 1. Three different solvents (benzene, methanol, and water) have been tested using this model. The introduction of the solvent results in a slight random increase of the free energy of reaction (ΔrG°) which does not depend on the dielectric constant.
By contrast, the increase of the dielectric constant leads to an increase in the free energy of activation (ΔG #) and the energy of activation (Ea). The calculated energy barrier for this reaction is therefore dielectric constant-dependent. This is in agreement with another theoretical study on a similar molecule. 53 In the case of benzimidazolthione, the effect of the solvent on the stabilization presents similar results to benzimidazolone. A theoretical study made on a similar compound supports this result. 46
This transition state reveals a pseudo-cycle of four atoms where the angle O1–C1–N1 changes in order to bring closer the two atoms and facilitate the transfer of the proton (see Figures 2 and 4). Even if the reaction appears to be not requiring much energy, as it is just a change in the angle H1–N1–C1, the activation barrier is too high.
It is known from the work of Polanyi and Zewail 54 that “the entire journey of a reaction, often involving a 10 configurational change, takes 1–10 ps.” Femtosecond dynamics is able to detect the evolution of a chemical reaction and the localization of the transition state. All experimental results fit well with theoretical results from Potentiel Energy Surface (PES) investigations. The interacting molecules will not follow the path of least energy provided by the IRC model, but they oscillate around this path.
It is fair to say that the optimized geometries at each point of the energy profile do not reflect those adopted experimentally, but they must not be too divergent because they show a continual transition from the situations of the reagents to those of the products. The first derivative of the energy profile (lower parts of Figure 3(a) and (b)) constitutes the reaction force. Three key points along an IRC have been highlighted by Toro-Labbé and co-workers. 55 and Politzer and co-workers.56,57 Using information from the IRC curve is a way of zooming in to glimpse and rationalize a reaction mechanism in terms accessible to any chemist.58–60
From the energetic profile associated with TSOG1 (Figure 3(a)), the entry of the hydrogen in the zone of influence of the TS was observed toward the coordinated (-1). The structure obtained is a quasi squared plane that involves a four-center reaction mechanism. Which represents the entry of the hydrogen in the zone of influence of the TS, the structure obtained is a quasi squared plane that involves a four-center reaction mechanism. Such situations are forbidden by the orbital symmetry rule known as the second rule of Wigner. 61 This explains why the energy barrier is very high. To overcome this prohibition, we have carried out calculations by imposing the assistance of a molecule of protic solvents.
Tautomerism process assisted by a molecule of a protic solvent
The uncatalyzed reaction may even be undetectable because the real reaction is catalyzed by KOH, 62 NaOH, 63 ice/CO2, 64 and tBuOH. 65
In order to take into account catalytic effects, we will begin with the action of a water molecule that effects a transfer of type (1,3). We optimized the geometry of the TS TSWO1 and verified by a vibrational analysis the presence of only one imaginary frequency.
To ensure that the TS connects the two minima WO1 and WO2, we calculated its energy profile (Figure 3(b)) as a function of the IRC.
We have found the expected compounds as minima on either side of the saddle point (Figure 3(b)). The optimized structures of ketone WO1, enol WO2, and the TS TSWO1 are presented in Figure 4. The addition of this molecule of water avoids the H1–N1–C1 angle constraint and allows the formation of a plane of six centers, with an angle of 124.49°.
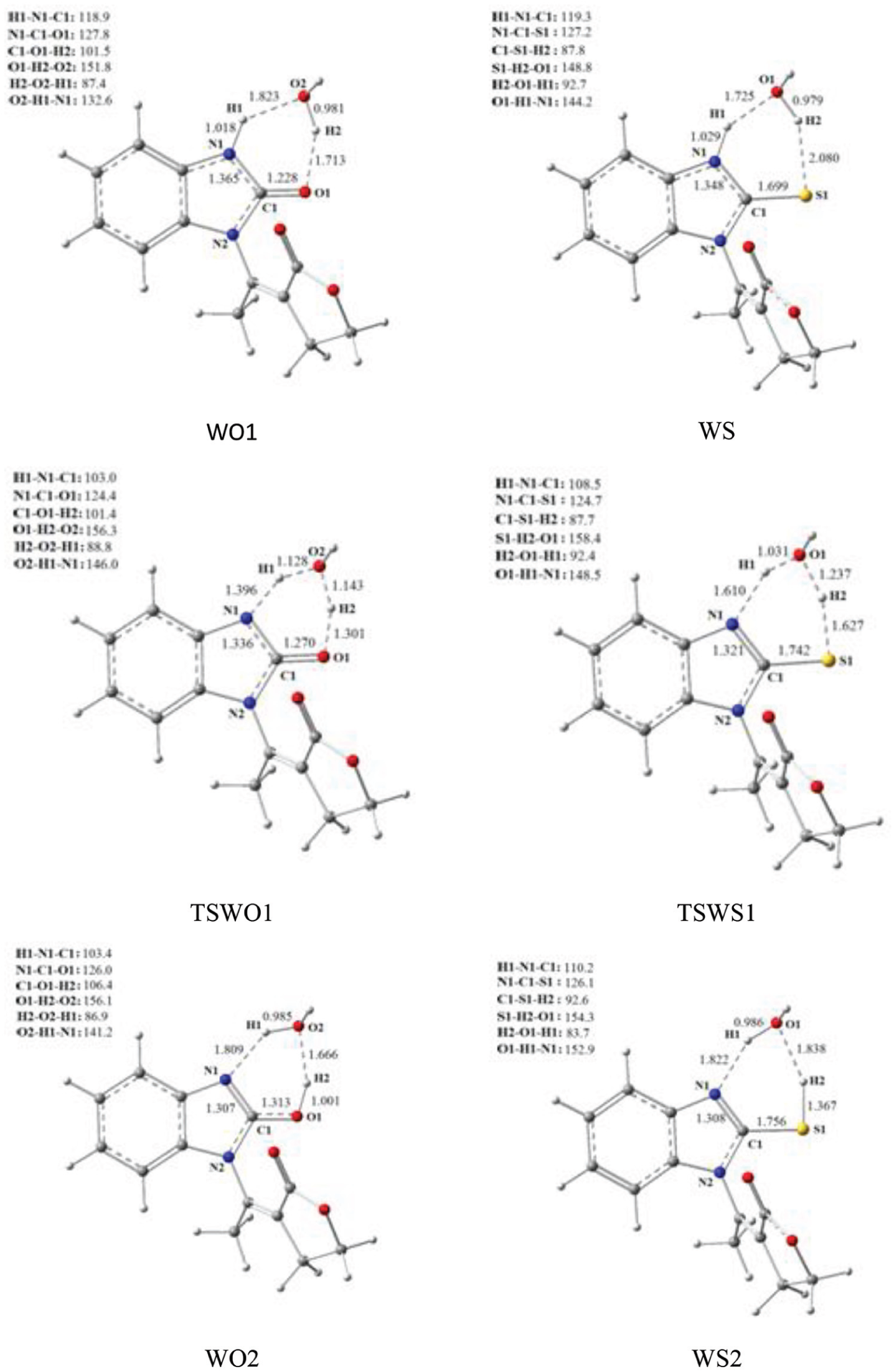
Optimized geometries involved in water-assisted tautomerism reactions.
The Gibbs energy ΔG of the proton transfer is decreased considerably to 10.83 kcal mol−1. As expected, the activation barrier by 18.70 kcal mol 21 . These values are similar to those of experimental studies.41,43,66
During tautomerism, H1 is transferred from N1 towards O1 and a bond is established between H2 and O1. In order to study the synchronicity of these different processes, the evolution of the distances C1–O1, N1–H1, O2–H1, O2–H2, and H2–O1 during this step has been analyzed. They are represented in Figure 5.
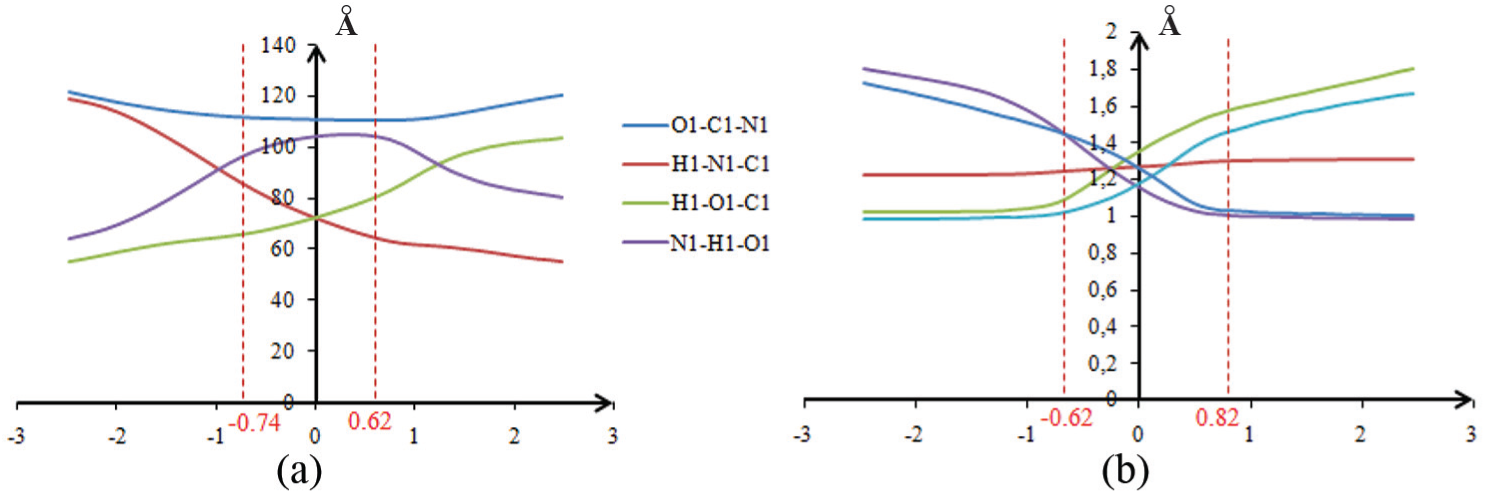
Evolution of (a) valence angles during proton transfer in benzimidazolone in the gas state and (b) internuclear distances during proton transfer in benzimidazolone assisted by a molecule of water.
In zone 1, the region of approaching reactants, the distances O2–H1 and H2–O1 decrease while the distances C1–O1, N1–H1, and O2–H2 remain substantially constant. The water molecule approaches the benzimidazolone tautomer in order to be correctly oriented and to allow the electronic transfers that will lead to the transfer of H1 as well as the creation of the bond O1–H2. In zone 2 which represents the TS zone of influence, the distances O2–H1 and H2–O1 continue to decrease until reaching the values 1.01 and 1.05 Å, respectively, while the distances C1–O1, N1–H1, and O2–H2 increase, indicating that the transfer of H1 is almost complete. In zone 4, the region of products formation and separation, the distances O2-H1 and H2-O1 stabilize.
However, the distances C1–O1, N1–H1, and O2–H2 continue to increase before being also stabilized, reflecting the formation of the H2–O1 bond. These results are supported by the work of Morell et al. 67 who suggested that the zone of the approach of reactants is located before the minimum response force while the products zone is after its maximum. Between these two points occur the electronic interactions responsible for the proton transfer and the formation of the bond H2–O1.
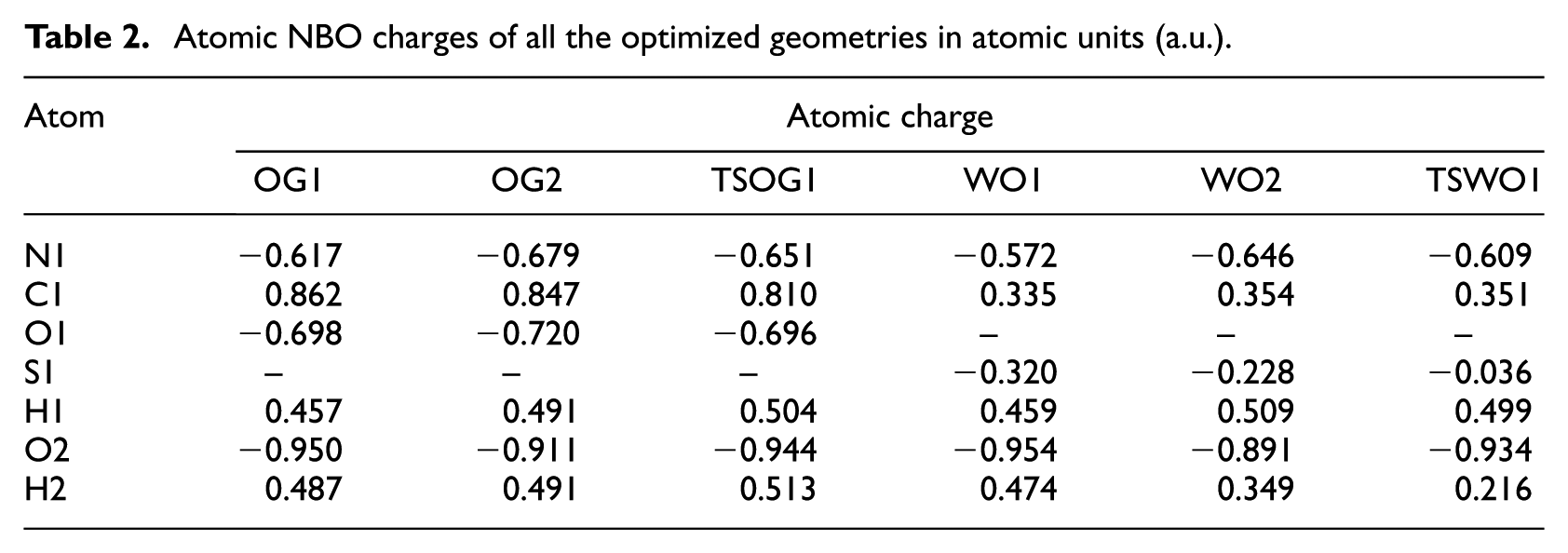
In order to analyze better these electronic interactions, we performed Natural Bond Orbital (NBO) analysis, which is a valuable tool for the study of bonding interactions, both intramolecular and intermolecular and for the transfer of charge in chemical compounds.68–70 The atomic NBO charges of all the optimized geometries are compiled in Table 2. These values indicate a high positive charge on the atoms H1 and H2 during the tautomerism. The negative charge of the oxygen atom acting as donor increases, while the charge of nitrogen which is the acceptor, decreases.
Atomic NBO charges of all the optimized geometries in atomic units (a.u.).
To generalize the study of the assistance process to other protic solvents, we focused on the tautomerism of benzimidazolone in the presence of a molecule of (C6H12) Cyclohexane, (CH3OH) Methanol, and (H2O2) Hydrogen Peroxide. This latter compound is not a usual solvent for such reactions. We studied it because it is a stronger (pKa = 11.7) acid than water (pKa = 15.7) and methanol (pKa = 15.5), in order to highlight better the role of mobile hydrogen. The corresponding values, compiled in Table 3, show similar results for the assistance by a molecule of methanol or hydrogen peroxide (H2O2) to that of water, but the presence of a C6H12 molecule presents gas state-like values. The structure of this transition state is not a six atom-ring but can be described as a five atom-rin containing two hydrogen neighbors and remains very close to the four-center TS geometry, which explains its high value. This observation highlights the role of polar protic solvents in the role of assistance aforementioned.
Thermodynamic and kinetic parameters in kcal mol−1 of benzimidazolone tautomeric reaction assisted by a protic solvent molecule.
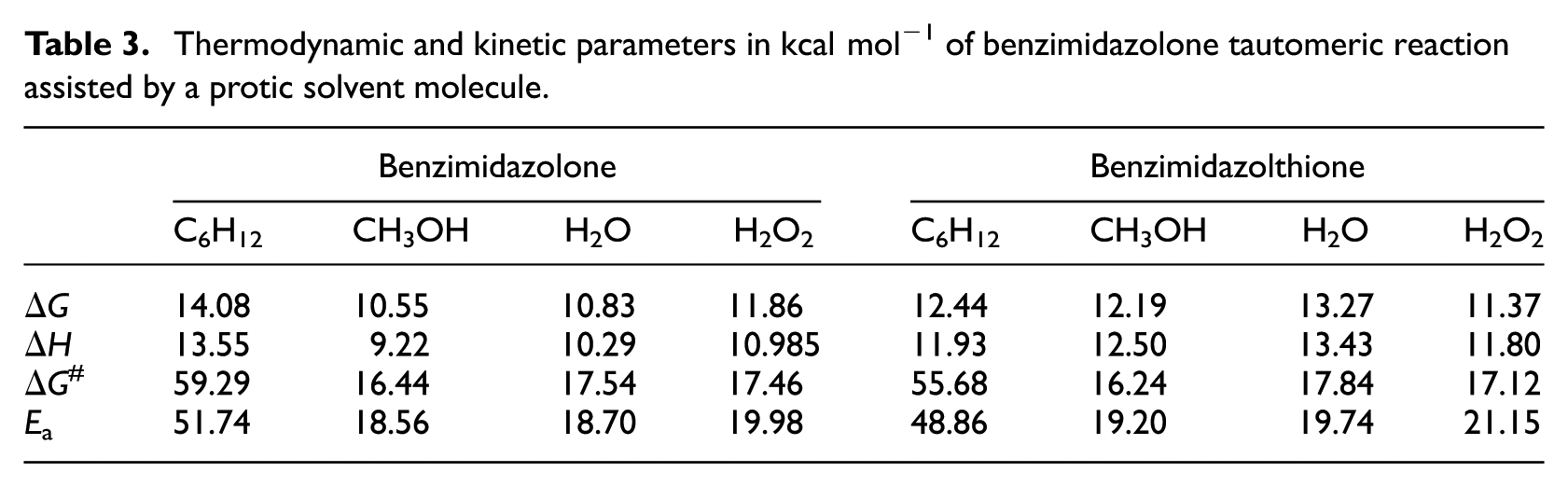
From a kinetic point of view, proton transfer assisted by methanol, water, or hydrogen peroxide leads to a decrease of activation energy barrier by about 34 kcal mol−1 as compared to proton intramolecular transfer in the gas phase. In addition, the molecule of methanol causes the most important decrease of barrier energy of 22.54 kcal mol−1. We can conclude that the methanol facilitates the exchange of hydrogen more than water and hydrogen peroxide.
Particular attention has been given to the geometry of the transition states TSMS1, TSWS1, and TSOS1 corresponding to the reactions of tautomerism of benzimidazolthione assisted by a molecule of methanol, water, and hydrogen peroxide, respectively (Figures S1 and S2 in the supplemental material) (see Figure 4).
Overall, the structures of these three TSs are similar. However, we note a few differences in distances between atoms. The distance between H1 and N1 is more important in TSMS1 than in TSWS1 and TSOS1. On the other hand, the distance between S1 and H2 is shorter in TSOS1 than in the other TSs. These results indicate that the rupture of bond N1–H1 is readily assisted by a molecule of methanol, whereas the transfer of hydrogen from peroxide to sulfur is more favorable.
In order to understand the difference between keto–enol and thio–enol tautomerism reactions assisted by methanol, water, and hydrogen peroxide, the profiles of reaction are shown in Figure 6.
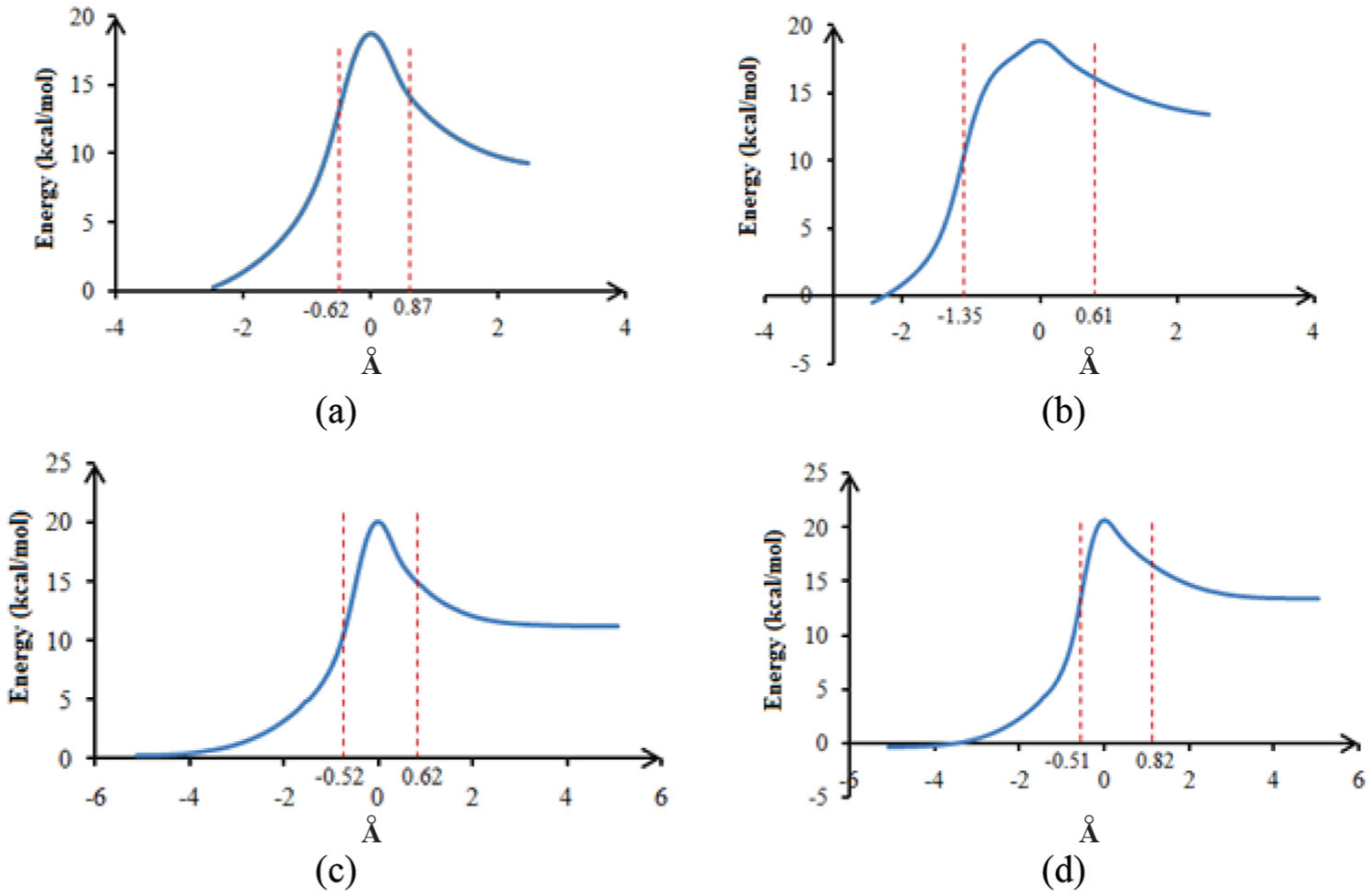
Energy profiles of benzimidazolone and benzimidazolthione tautomerism assisted by H2O for (a) and (b) and by H2O2 for (c) and (d).
We note that the TS zones of influence in methanol, water, and hydrogen peroxide are respectively equal to 1.54, 1.49, and 1.14 (Å) in the keto–enol tautomerism reaction and 2.09, 1.96, and 1.33 au in the thio–enol tautomerism reaction. This indicates that there is a large geometrical variation in the assistance with methanol than with the two other molecules.
The potential energy profiles of keto–enol and thio–enol tautomerism assisted by methanol and water are also very different. In fact, the shoulder of the potential energy before the transition state for thio–enol tautomerism is absent in the profile of the reactions of keto–enol tautomerism. It is a direct consequence of the asynchronous nature of the various processes that take place during thio–enol tautomerism.
In order to check this, the evolution of selected distances highly modified during tautomerism has been studied (Figure 7). We find in the two reactions assisted by methanol and water that the distance N1–H1 increases strongly before the transition state, whereas the distance O1–H2 increases after it. This indicates that the process is clearly asynchronous in contrast to that observed in the case of keto–enol tautomerism assisted by a water molecule, previously described.
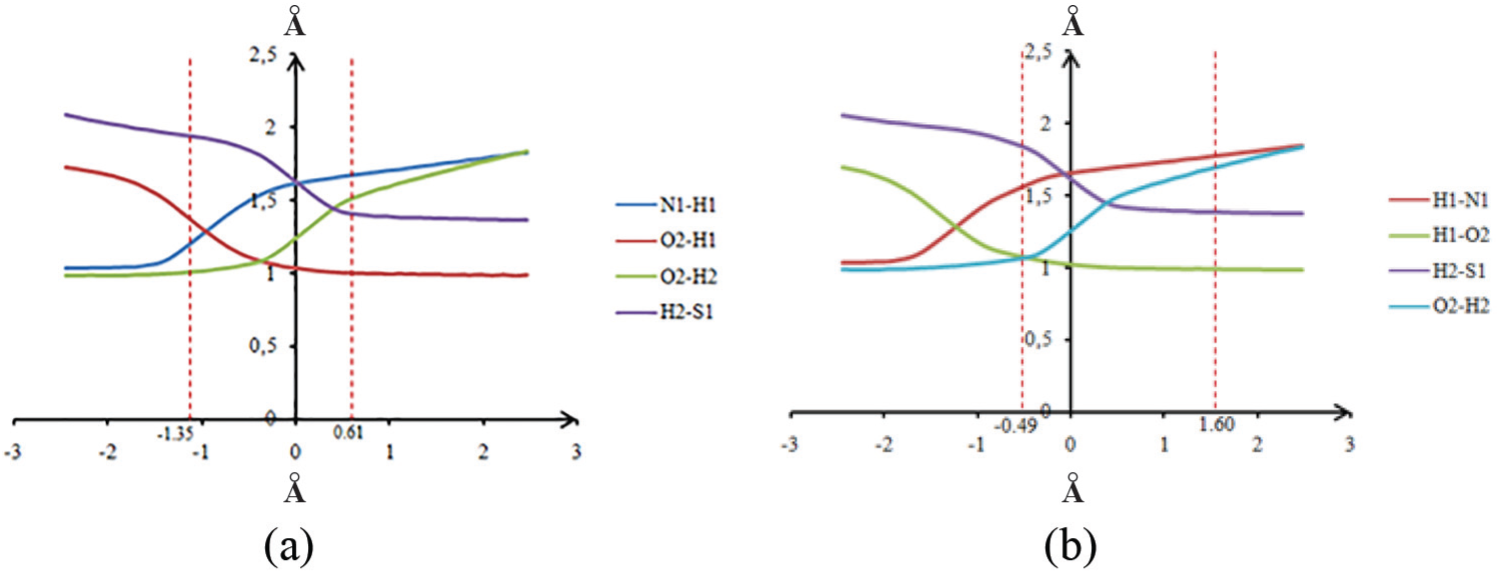
Evaluation of internuclear distances in benzimidazolthione: (a) assisted by a molecule of water and (b) assisted by methanol.
Conclusions
Here we have presented the results of theoretical studies of keto–enol and thio–enol tautomerism reactions in benzimidazolone and benzimidazolthione using the Cam-B3LYP functional together with the 6-311G(d,p) basis set. The effect of a molecule of polar protic solvent on proton transfer reactions has been studied with three different solvents (methanol, water, and hydrogen peroxide) using the PCM model. The structural parameters of benzimidazolone and benzimidazolthione molecules were calculated in the gas phase and in the solvents. The results are strongly in agreement with the experimental data of X-ray diffraction of similar molecules. The noted energy barriers for the keto–enol and thione–thiol tautomerism in the gas phase and in solution are very high, indicating disadvantageous processes. The assistance by a solvent molecule (methanol, water, and hydrogen peroxide) stabilizes the ketone and thione forms. Calculated results show that the assistance by a molecule of methanol, water, or hydrogen peroxide considerably reduces the activation barrier of keto–enol and thio–enol tautomerism reactions. In addition, the molecule of methanol effects the greatest decrease of the energy barrier. The main difference presented by the thiol compound is the asynchronism detected in the tautomerization process.
Supplemental Material
PRK825740_ESI_v2 – Supplemental material for Proton transfer in the benzimidazolone and benzimidazolthione tautomerism process catalyzed by polar protic solvents
Supplemental material, PRK825740_ESI_v2 for Proton transfer in the benzimidazolone and benzimidazolthione tautomerism process catalyzed by polar protic solvents by Hasnia Abdeldjebar, Yamina Belmiloud, Wassila Djitli, Sofien Achour, Meziane Brahimi and Bahoueddine Tangour in Progress in Reaction Kinetics and Mechanism
Footnotes
Acknowledgements
This work is the result of Algerian and Tunisian teams cooperation. The authors are very grateful to the Laboratory of Theoretical Physical Chemistry and Computational Chemistry of Chemistry Faculty of U.S.T.H.B. They specially thank Tunis University, El Manar for hosting their students in training, in particular Professor Bahoueddine Tangour and his team for all their valuable support.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.
Supplemental material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.