
Abstract
In this work, we focus our attention on chemical reactions confined within carbon nanotubes. As a result of the confinement within carbon nanotubes, novel physical and chemical properties are found for the confined materials. We consider the feasibility of proton transfer inside carbon nanotubes. To do that, we have chosen formamide as the simplest real model for exhibiting the tautomerization in DNA. We have used the quantitative structure–property relationship method, based on geometry optimization and quantum chemical structural descriptors, to illustrate the potential of using the confined space inside carbon nanotubes, which will provide comprehensive information about carbon nanotubes. All calculations have been carried out using density functional theory quantum calculations with the B3LYP functional. The geometries optimized by the Gaussian program were transferred to the computer software DRAGON to calculate pertinent descriptors that could be used in the quantitative structure–property relationship model.
Keywords
Introduction
One of the principal objectives of the chemist is to create and control new chemical reaction processes. Several methods have been developed to understand these pathways and therefore our ability to improve different chemical transformations.
Recently one of the adopted tools, which has attracted increasing attention, is the confinement of guest species within a nanotube. In this work, we will concentrate on the chemical reactions confined within carbon nanotubes (CNTs).1–7
Exploration of the free space inside a CNT and our understanding of the different phenomena that can occur them within have been examined by several theoretical and experimental works on different probes. These studies suggest that molecules confined within CNTs display different properties from the corresponding isolated systems. This difference depends on the diameter of the CNT.8–12
CNTs can be used for therapeutic purposes and have properties that make them an excellent tool in biomedical applications. However, we note that the cytotoxicity of CNTs can influence their usage in biological systems.13–16 CNTs have excellent sites for physisorption phenomena because of the potential gaps in their interior.17–20
The CNT diameter is one of the most important parameters in determining the ability to trap and control a molecule inside a CNT. Thus, it has been assumed that the use of nanotubes as boxes or cages can ensure the transport of the drugs. Studies21–24 indicate that the CNT diameter is one critical condition for the encapsulation of the anticancer drug cisplatin.
Other works show that the location of a molecule inside a CNT has a significant effect on the interaction between the molecule and the wall of the tube.25–27 We consider here the feasibility of proton transfer (PT) within CNTs using theoretical calculations. We have used small-sized molecules as an example of this type of chemical reaction. PT and proton coupled electron transfer reactions are important processes in enzymatic catalysis mechanisms.28–33
In this study, we were interested in the potential actions of a CNT in the transport of formamide (FA) to a target such as DNA. We note that the FA content plays a very important role in the assembly of DNA, and it is among the factors influencing the molecular hybridization of DNA. Indeed, the FA creates hydrogen bonds with the nitrogen bases of the DNA molecule and the denaturation will be faster. The FA will also tend to destabilize imperfect hybrids.34,35 Such a chemical process requires the computation of a potential energy surface (PES). 36 Generally, PT processes occur in solution, as is relevant to biological effects. 37 Recently, PT reactions in acid–base complexes and solute–solvent systems became of interest due to their major biological importance.38–44
The theoretical study summarized in our work demonstrates how the pathway of the PT reaction can be influenced by confinement in a CNT. However, there are some questions about the energetics of compounds inside the CNT. For example, the characteristics of a CNT are dependent on the tube diameter. It is not clear how the size and chirality of the CNT affect the PT reaction at the energetic and geometric levels. We have used the quantitative structure–property relationship (QSPR) method,45,46 which is based on geometry optimization at the level of density functional theory (DFT) 47 and quantum chemical structural descriptors, to illustrate the potential of using the confined space inside CNTs to dictate the outcome of chemical reactions, which will help us to understand the effect for this type of reaction and will also provide comprehensive information about the CNT. Consequently, we chose four CNT types of equal length and different diameters. We focus on the following: the FA–FI (where FI is formamidic acid) tautomerization confined within CNT(5,5), CNT(6,6), CNT(7,7), and CNT(8,8). The examples illustrated here compare the effects of confinement on the molecules. We applied the DFT method to localize the stationary points for reactants, transition states (TSs), and products on the PES.
Computational methods
All DFT calculations were performed with the Gaussian 09 program package 48 using the hybrid functional B3LYP together with the 6-31G* basis set.47,49 For the molecules confined inside CNTs, full geometry optimizations were performed considering mechanical and electrostatic embedding with the two-layered ONIOM (our own n-layered integrated molecular orbital and molecular mechanics) approach.50,51 As a first step, the CNT was put into the lower layer using the universal field force (UFF). 52 For any TS, one imaginary frequency was observed. Vibrational frequency calculations were used to determine the zero-point energy corrections. We have calculated the basis set superposition error (BSSE) corrections to obtain the corrected interaction energies using the counterpoise correction procedure.53,54
To estimate the effect of solvation, we have chosen the integral equation formalism of the polarizable continuum model (IEF-PCM)55,56 at the same level of theory as the gas phase calculations with the dielectric constant that corresponds to the aqueous solvent (H2O, εr = 78.3).
We have transferred all geometries optimized by the Gaussian program 48 to the computer software DRAGON. 57 We have eliminated all descriptors with constant values (the standard deviation is practically negligible) and those that are highly correlated.
Among these descriptors, we have chosen the very important ones which correspond to the molecular volume or the van der Waals (vdW) volume. The important descriptors were then selected by the genetic algorithm, in the version of MobyDigs software due to Todeschini et al. 58
Results and discussion
Energetic stability and geometrical parameters
In this part, we will look at the possibility of finding a similar confined structure to the isolated structure, but with lower energy. This means to find a structure that has different properties.
The length of the CNT segment which has been used in this work is 17.5 Å, namely: C140H20 for CNT(5,5), C168H24 for CNT(6,6), C196H28 for CNT(7,7), and C224H32 for CNT(8,8) (Figure 1).
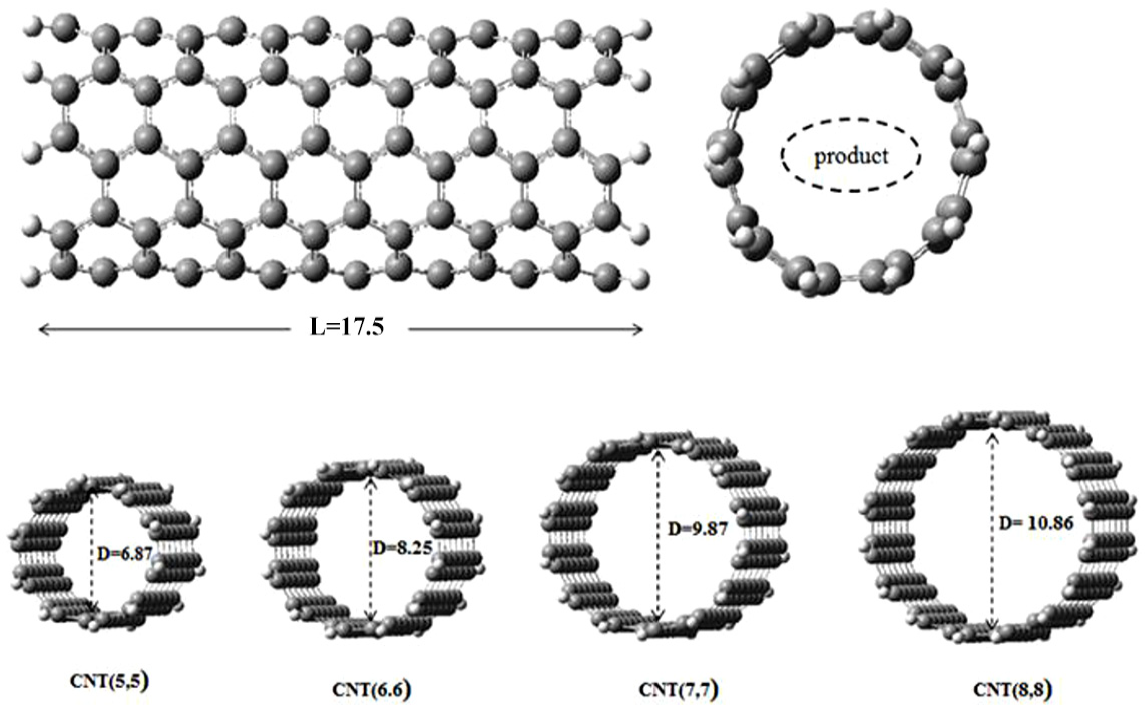
Various sizes of carbon nanotubes used.
The molecular structures of FA, FI, and TS are shown in Figure 2.
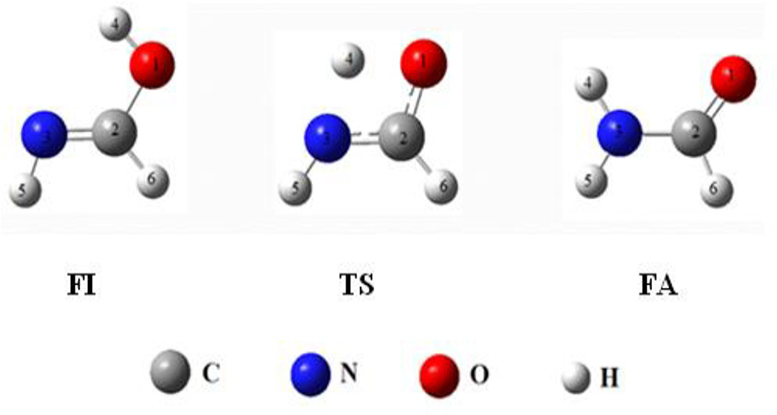
Schematic representation of products and TS computed at the B3LYP/6-31G* level.
The hydrogen atom which is transported is atom H4. This is because, in the PT reaction on the H5 atom, the rotation around the double bond C=N requires more energy, and also there is repulsion between the hydrogen atoms (Figure 2).
Because of the narrow inner space of the CNT, we can find some molecules in different configurations; this can be seen from some changes of the geometric parameters. The most important changes are observed in the CNT(5,5) case.
Regarding the FI structure: the NCO bond angle of FI inside CNT(5,5) (125.8°, Table 1) is larger by about 4° compared to the other FI structures (confined and isolated). The smallest value for angle O1C2H6 is observed in CNT(5,5) (108.7°).
Structural parameters for FA, FI, and TS at the B3LYP/6-31G* level.
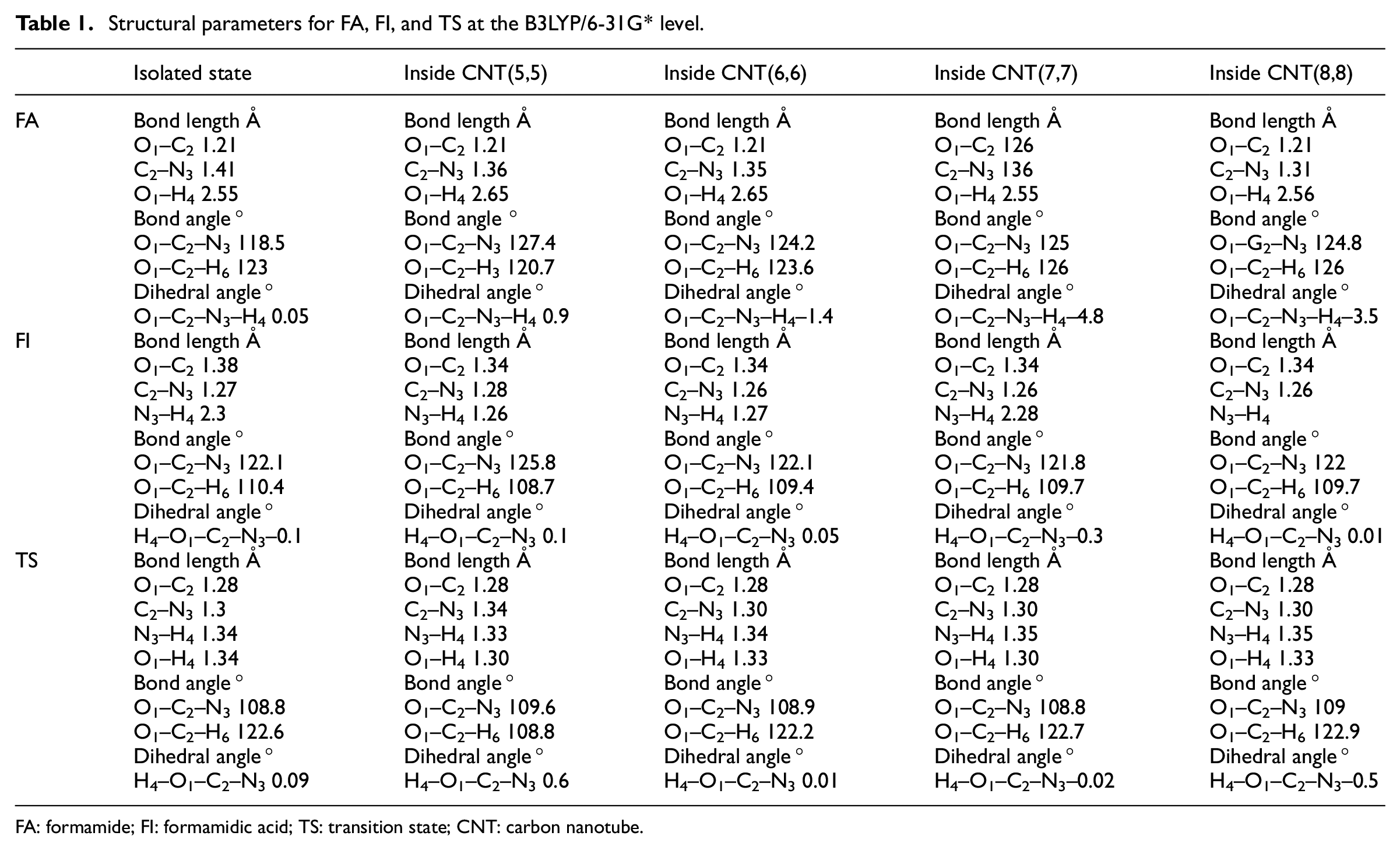
FA: formamide; FI: formamidic acid; TS: transition state; CNT: carbon nanotube.
Regarding the FA structure: it can be seen that the C–N bond length in the FA confined into CNTs is shorter by 0.05 Å compared with the isolated state. The largest NCO angle is observed in CNT(5,5) (127.4°). The O1C2H6 bond angle is reduced to 120.7°; we see that the O1–H4 distance in the FA inside CNTs is longer by 0.1 Å compared with the isolated FA.
Regarding the TS structure: the O1–H4 distance in the TS (1.30 Å) is decreased by the confinement and the C–N bond length is longer by 0.04 Å than in the isolated state. The O1C2H6 angle of the TS in CNT(5,5) is smaller by about 13° compared with the isolated state and other confined CNTs. Thus, the structure of the TS is affected by the confinement of CNT(5,5). This effect is evident at the energy levels.
It is clear that confinement energy and the product–CNT interactions 21 are the important factors to stabilize the products inside the CNT. Based on this, the relative importance of these interactions on the guest species within the CNT can be significantly affected by the change in morphology of each system studied; in principle, it could also lead to the changing of the activation barrier. To illustrate the actual effect of the tube size on the energetics of reaction, we examined various CNT model systems with different diameters. The confinement energies (Econf) of placing FA and FI into CNTs are exothermic and are listed in Table 2. The Econf for all the geometries is calculated using DFT/B3LYP by the formula

Confinement and BSSE energies (kcal mol−1) of FA and FI inside CNTs computed at the B3LYP/6-31G* level.
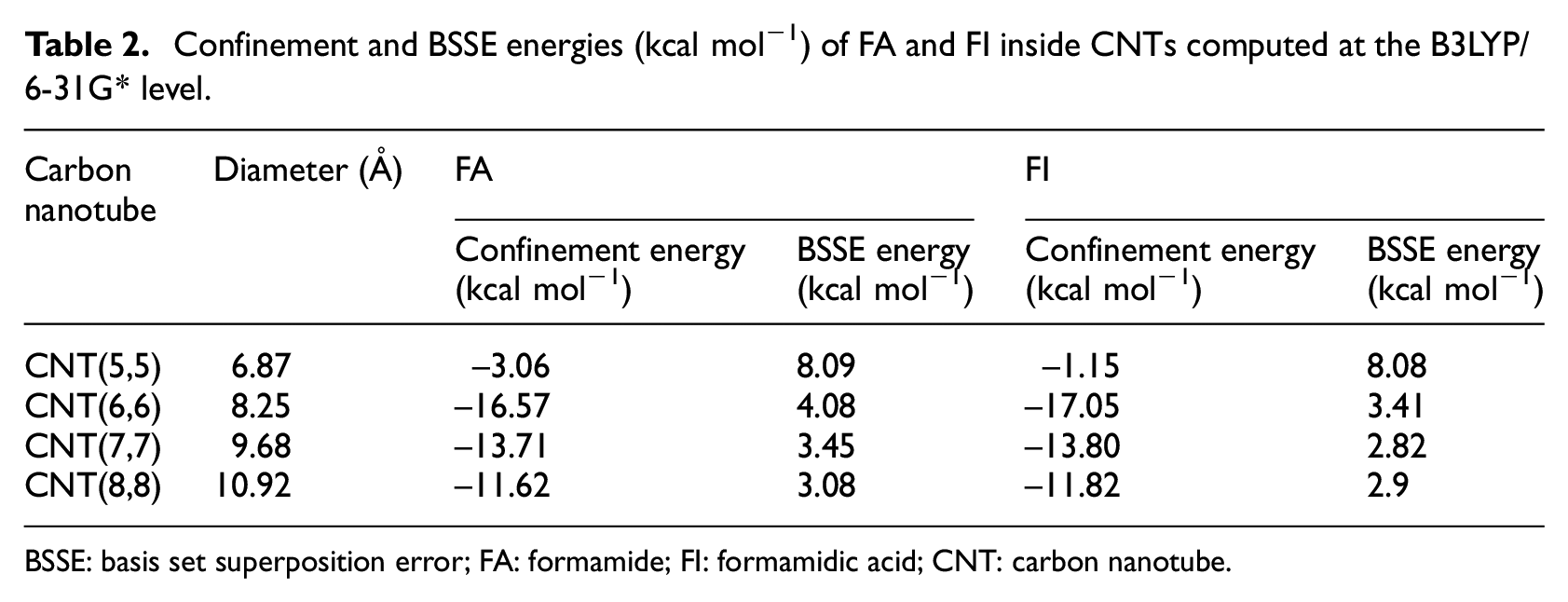
BSSE: basis set superposition error; FA: formamide; FI: formamidic acid; CNT: carbon nanotube.
We note that the difference between the results indicates the important interactions in the course of the nano-confinement process. The efficiency of the non-bonded interactions between the reacting system (FA, FI) and the electronic density of the CNT can be explained by the structural changes of the geometry of the TSs. In our systems, the bonds O–H, N–H, and C–O are polarized, but the C–H bond is non-polar. In a polar bond, the excess negative charge is associated with the electronegativity of the oxygen and nitrogen atoms. The carbon atoms of the CNT can interact with the C–H, C–N, and C–O bonds via some interaction such as C–H…π, C–O…π, and C–N…π.
These values of Econf are generally considered as an upper limit for the physical interactions of vdW type. The confinement energies related to placing FA (–16.57 kcal mol−1) and FI (–17.05 kcal mol−1) into CNT(6,6) are larger than those for placing FA and FI into CNT(5,5), CNT(7,7), and CNT(8,8). Thus, the most important difference of energy is found for CNT(6,6) (see Table 2).
Where the diameter of the nanotubes increases, Econf decreases by about 2.86 kcal mol−1 (FA inside CNT(6,6) → FA inside CNT(7,7)) and 4.95 kcal mol−1 (FA inside CNT(6,6) → FA inside CNT(8,8)), respectively. The Econf decreases by about 3.25 kcal mol−1 (FI inside CNT(6,6) → FI inside CNT(7,7)) and 5.23 kcal mol−1 (FI inside CNT(6,6) → FI inside CNT(8,8)), respectively.
When the tube is narrowed to 6.87 Å diameter (CNT(5,5)), the energy is drastically reduced. The Econf of FA and FI are −3.06 and −1.15 kcal mol−1, respectively. For less than this diameter, we found an endothermic process.
These different results implicate the key role of the vdW-type interactions. The values of BSSE are very low (see Table 2), indicating the appropriate basis set used to study the complex system. This could signify that it does not affect the final results. The activation energies and the relative energies for PT reaction processes are presented in Table 3. All energy values include zero-point vibrational energy corrections. There is a correlation between the confinement energy (Table 2) and activation energy (Table 3).
Relative and activation energies of PT reaction in confined and isolated states at the B3LYP/6-31G* level (shown in parentheses are the values obtained for the polarizable continuum model (IEF-PCM)).
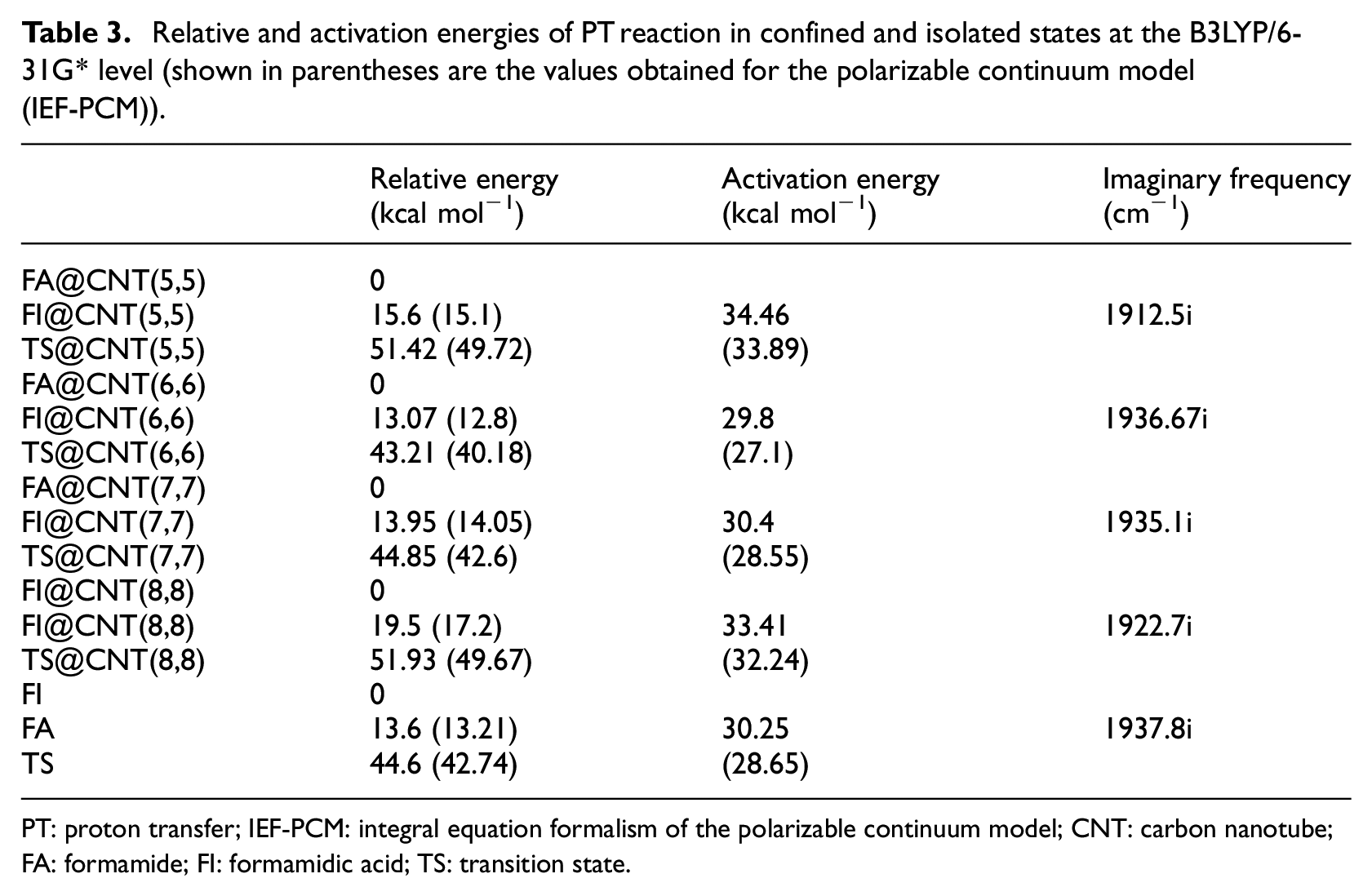
PT: proton transfer; IEF-PCM: integral equation formalism of the polarizable continuum model; CNT: carbon nanotube; FA: formamide; FI: formamidic acid; TS: transition state.
We can see that the energy of the TS inside CNT(6,6) is 43.21 kcal mol-1. The lowest activation energy (Ea = 29.8 kcal mol-1) corresponds to the confinement in CNT(6,6) and its stability which is determined by the relative energy values (see Table 3).
Product FA becomes more stable under the confined CNT(6,6) state because the energy calculated is lower than that of the other FAs (13.07 kcal mol−1) (Table 3). Larger values of Ea are observed for the larger tube CNT(8,8) (33.41 kcal mol−1) and the narrowest tube CNT(5,5) (34.46 kcal mol−1) (see Figure 3).
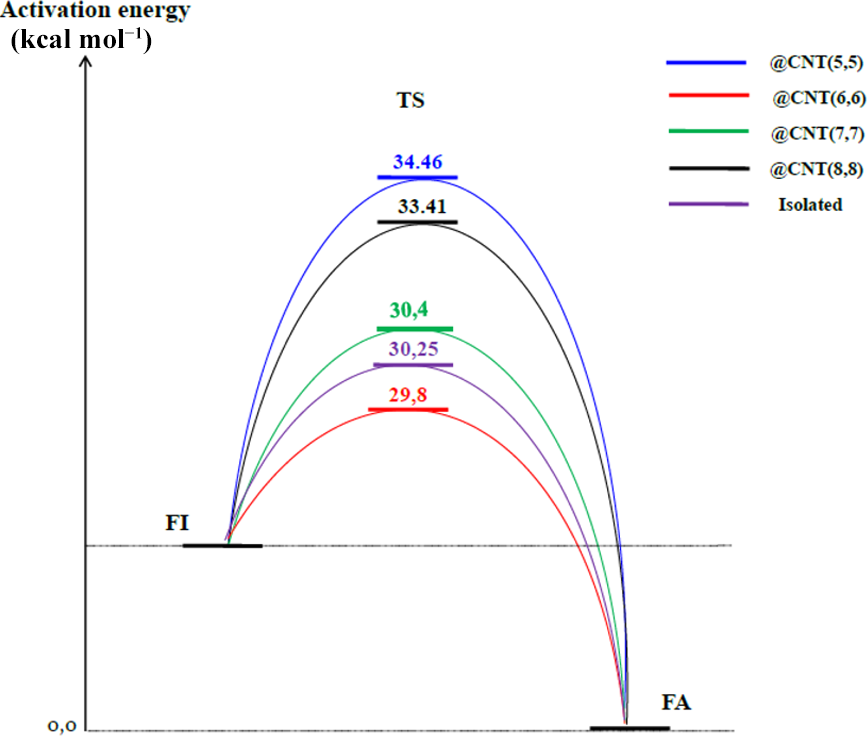
Activation energies of PT reaction in the isolated and confinement states.
The effect of water as the solvent was studied using the IEF-PCM model. We observed that the energy barrier decreases in each of the confined and isolated systems. For example, in CNT(6,6) the energy barrier decreases by about 3 kcal mol−1 (Table 3). These energy barriers are influenced by the polarity of the water solvent.
Obviously, the reacting system is affected by tube diameter and the consequent changing volume inside the CNT. According to Williams et al., 59 the degree of overlap between the vdW radii of two species causes the attraction or repulsion between guest species, which leads to stabilization or destabilization of the system. Thus, this result supports our idea for this type of interaction.
We observed that the confinement of FA and FI within CNTs with different dimensions produces several phenomena that can affect the interactions with the CNT walls. Because of the short distances between the hydrogen atoms of our molecules and the tube wall (CNT(5,5)), this implies C–H…π repulsive interactions that cause a destabilization of the TS and thus lead to a large activation barrier (34.46 kcal mol−1). Clearly, narrowing the CNT gap reduces the size of the molecule in the TS. Thus, there is a large overlap between the vdW radii of the atoms. The access of the C–H bonds in the TS to the CNT wall is more difficult than for FA and FI when the CNT gap is narrower. So, we can explain that the oxygen, nitrogen, and especially hydrogen atoms will interact directly with the CNT wall (larger overlaps of vdW volume), which affects the change of the structural (e.g. volume) and energetic values. Wider tubes like CNT(8,8) are characterized by the absence of a significant effect of the nano-confinement. The Ea is equal to 33.41 kcal mol−1 (Figure 3) in comparison to the isolated system (30.25 kcal mol−1). Because of that, it is better to consider again this type of interaction as the most important factor to stabilize guest species inside CNTs and to understand the variation of the energy barrier.
Molecular descriptors
In order to determine the properties of molecules inside CNTs with different sizes, the QSPR approach 45 can be used to predict the vdW volume for each molecule. The main goal of any QSPR study is to establish correlation models based on the molecular descriptors of molecular structure and chemical databases which allows defining the determining factors for the measured property.60–62
The purpose of this study was to create a mathematical model by multiple linear regression (MLR) analysis which allows us to connect the physical chemistry properties of the molecular descriptors calculated by DRAGON 57 according to the different sizes of the CNTs.
By performing the correlation between the descriptors used according to the vdW volume of our molecular atoms (three-dimensional (3D) molecular representation of structure based on electron diffraction weighted by atomic vdW volumes)62,63 and the set of CNT sizes, we have to use the following equation with an MLR analysis: Y = b0 + b1 × X1 + b2 × X2, where Y is the response or dependent variable, X is the independent variable and bi (i = 0, 1, 2) is the regression coefficient associated with it.
After regression analysis, we have selected two molecular descriptors from the appropriate models obtained to establish MLR models. 3D-MoRSE (3D molecular representations of structure based on electron diffraction) type descriptors were formulated by Schuur and Gasteiger in 1996.64,65
Some studies utilizing 3D-MoRSE confirmed that the separation of molecules according to their structural properties is mainly dependent on the distances between the atoms.66,67 One of the proposed types concerns the vdW volume such as Mor19v (3D-MoRSE—signal 19/weighted by atomic vdW volumes).
The MLR equations for the Ea of the PT reaction model in terms of two descriptors (diameter (D) and Mor19v) are
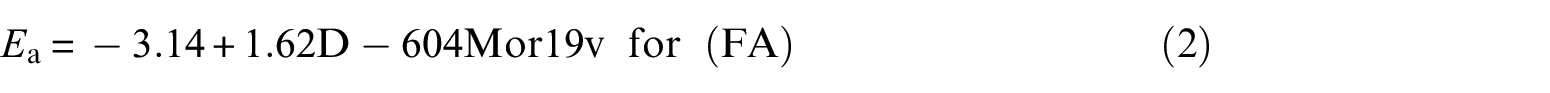

In order to find more reliable results, one should consider more than four points. The statistical parameters are reported in Table 4.
Statistics summary for the one-dimensional calculated models.
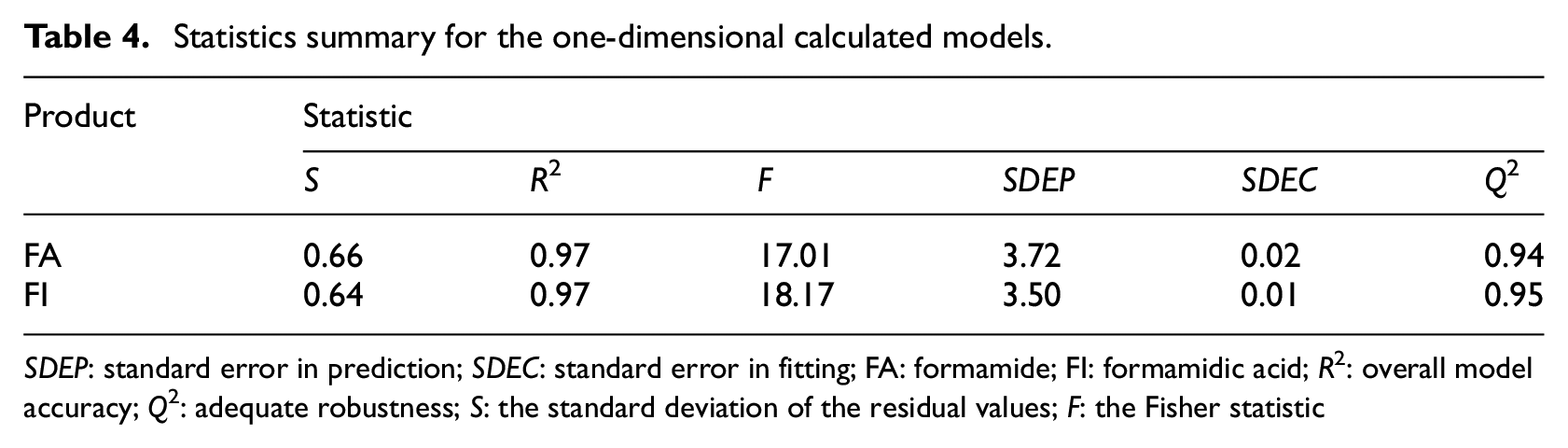
SDEP: standard error in prediction; SDEC: standard error in fitting; FA: formamide; FI: formamidic acid; R2: overall model accuracy; Q2: adequate robustness; S: the standard deviation of the residual values; F: the Fisher statistic
As shown in Table 4, the high values of the correlation coefficient R2 (0.97, 0.97) and adequate robustness Q2 (0.94, 0.95) for FA and FI, respectively, indicate good internal fitting ability and predictive ability in this model.
When the value of Q2 is greater than 50%, we will have a good model. In our study, we found that Q2 is greater than 90%, so we have an excellent model. 68
The quality of fit of the calculated models was evaluated by means of the multiple determination coefficients, where SDEP and SDEC are the standard error in prediction and standard error in fitting, respectively. 69
The model is robust; thus, the difference between R2 and Q2 is small (2.7% and 2.5% with respect to FA and FI). The model shows a good stability in internal validation.
Conclusion
In this study, we investigated, using DFT, the PT reaction inside CNTs with different diameters: (5,5), (6,6), (7,7), and (8,8), focusing on the effect of CNT diameter on the energetic stability and the geometrical parameters of the confined molecules. The interaction between the guest confined molecules and the nanotube is discussed. The C–H…π, C–O…π, and C–N…π interaction and vdW forces have an important role for the stability of the molecules inside CNTs; the narrowing of the gap of a CNT leads to a large overlap between vdW radii of the atoms. The appropriate diameter should be taken into consideration for achieving higher stability of the system. The lowest observed energy value is in the nanotube (6,6) due to this type of interaction. In order to have more reliable results, we developed QSPR models for several geometrical properties of a series of CNTs related to their chemical reactivity. The MLR model obtained in this study has very good fitting ability and acceptable predictive power and stability.
Footnotes
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was financially supported by the Ministére de l’Enseignement Supérieur et de la Recherche Scientifique (M.E.S.R.S) of Algeria, with PRFU project N° B00L01UN160420190019.