
Abstract
Density functional theory calculations were employed to investigate the electrochemical oxygen reduction reaction on the (111) and (100) surfaces of cobalt(II) oxide. Different mechanisms were applied to evaluate the oxygen reduction reaction performance of cobalt(II) oxide structures in terms of the Gibbs free energy and density of states. A variety of intermediate structures based on associative and dissociative mechanisms were constructed and optimized. As a result, we estimated the catalytic activity by calculating the free energy of the intermediates and constructing free energy diagrams, which suggested that the oxygen reduction reaction Gibbs free energy on cobalt(II) oxide (111) and (100) surfaces based on the associative mechanism is smaller than that based on the dissociative mechanism, demonstrating that the associative mechanism should be more likely to be the oxygen reduction reaction pathway. Moreover, the theoretical oxygen reduction reaction activity on the cobalt(II) oxide (111) surface was found to be higher than that on the cobalt(II) oxide (100) surface. These results shed light on the rational design of high-performance cobalt(II) oxide oxygen reduction reaction catalysts.
Keywords
Introduction
The unprecedented energy crisis and environmental pollution have focused global attention on next-generation energy devices like metal–air batteries and fuel cells.1–3 Among these, fuel cells have been widely accepted as a promising alternative due to their high conversion efficiency, high power density and low pollution nature.4–6 As a crucial part of fuel cells, the oxygen reduction reaction (ORR) electrode has attracted worldwide attention to pursue cost-effective, catalytically active, and highly stable materials for ORR catalysis.7–14 However, rational design of a high performance ORR catalyst based on theoretical understanding of oxygen reduction mechanisms still remains a great challenge.
An ideal electrocatalyst for the four-electron oxygen reduction should be able to facilitate the reduction process below the equilibrium potential of 1.23 V. 15 In the case of oxygen reduction to H2O, all the four charge-transfer steps should have reaction free energies of the same magnitude equal to the equilibrium potential of 1.23 eV.16,17 Therefore, noble metals (e.g. Pt) and their alloys are widely accepted as state-of-the-art ORR electrocatalysts.18–21 However, their limited reserves and cost have greatly limited their development for the ORR and prevent their large-scale application. 22 Recent experimental and theoretical studies have shown that earth-abundant transition metals (like Co) and their oxides act as promising ORR electrode materials with considerable intrinsic activity.23–27 For instance, Qiao’s group reported that single-crystal cobalt(II) oxide (CoO) nanorods with oxygen vacancies have very good ORR activity, 23 and the high-energy interfacial structure composed of CoO/Mn3O4 has better ORR performance than that composed of Pt/C. 28
However, deeper insight into the role of the CoO surface structure in the process of the ORR, ulitizing density functional theory (DFT) and thus providing guidance for enhancement of the ORR activity, remains a challenge to a fundamental understanding of the catalytic performance.29–32 Li et al. 33 have considered the ORR at the (111) face of CeO2 using DFT.
In this study, we focus on understanding the ORR process on the CoO structure using the DFT + U calculation method. The ORR on the different CoO surfaces was investigated, and the free energy of the intermediates was calculated based on two different mechanisms, namely, an associative mechanism and a dissociative mechanism. After studying the surface free energy diagrams, it was found that the overpotentials on both the CoO (111) and (100) surfaces based on the associative mechanism were lower than those based on the dissociative mechanism, indicating an easier ORR pathway. 34 Furthermore, the ORR Gibbs free energy on the CoO (111) surface was found to be smaller than that on the CoO (100) surface, suggesting a higher intrinsic activity on the CoO (111) surface. This study provides a theoretical basis for the ORR energetics on CoO surface structures, thus opening a new door for subsequent enhancement of their ORR activity.
Computational models and theoretical methods
For all calculations, the DFT level of theory was chosen for use with the Vienna ab initio simulation package (VASP).35,36 The projector-augmented plane-wave (PAW)37,38 method for describing electron–ion interactions, together with the spin-polarized generalized gradient approximation (GGA) of Perdew–Burke–Ernzerhof (PBE) type were employed to treat the exchange–correlation energy density functional. 39 The PBE exchange–correlation functional with the on-site Coulomb repulsion U = 3.50 eV40,41 was used. A full optimization of all the atom positions in the bulk CoO was performed via the action of a conjugate gradient optimization procedure. The Brillouin zone integrations were performed using a 5 × 5 × 5 Monkhorst–Pack grid for the bulk. A spin-unrestricted approach was adopted. To solve the self-consistent field convergence of the Kohn–Sham equations, the energy change convergence criterion was set at 1 × 10−5 eV between two successive iterations. In the bulk calculations, the positions of all ions were relaxed, and the cut-off energy was 400 eV.
To study the ORR catalytic activity on different CoO surfaces, we first sought to find the lattice parameters of preliminary CoO (111) and (100) surface models and confirm the optimized parameters for these surface models; the eventual established CoO (111) and CoO (100) crystal plane models are shown in Figure 1. The CoO (111) surface structure model is the unit cell with parameters a = b = 5.97 Å, c = 8.06 Å, α = β = 90°, γ = 120°, comprising a total of eight atomic layers, wherein the total number of atoms is 32 (Co 16 atoms, O 16 atoms). During the structure relaxation, the top four layers were relaxed, and the next four lower layers were kept frozen. The CoO (100) surface structure model is the unit cell with parameters a = b = c = 4.22 Å, α = β = γ = 90°, comprising a total of six atomic layers, the total number of atoms is 24 (Co 12 atoms, O 12 atoms). During the structure relaxation, the top three layers were relaxed, and the next three lower layers were kept frozen. In order to isolate the upper and lower supercells without them affecting each other, the periodic images of the surface were separated by at least 20 Å of vacuum. 42 The first Brillouin zone was sampled using a 5 × 5 × 1 Monkhorst–Pack grid of special k-points for the slab calculations.15,27
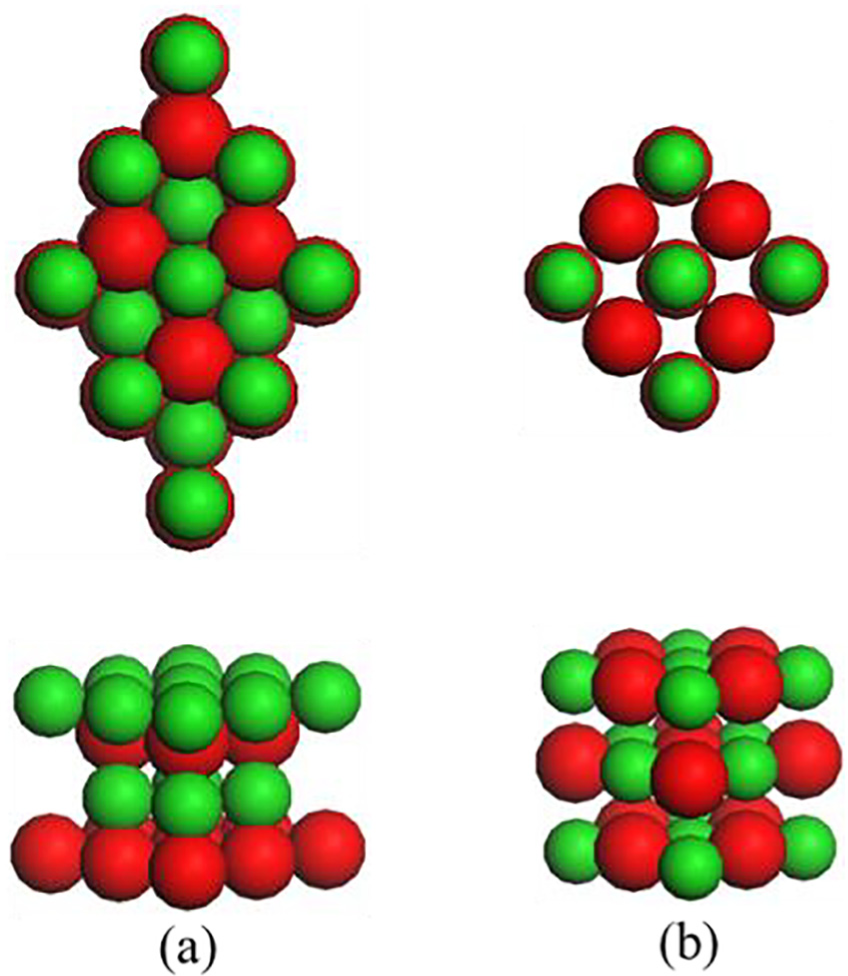
Structure models of (a) CoO (111) surface and (b) CoO (100) surface. Top/bottom rows are the top/side views of the model. Co and O are coloured in green and red, respectively.
Results and discussion
The overall reaction scheme of O2 reduction to H2O is: O2(g) + 4H+ + 4e– ⇌ 2H2O(l) with two possible reaction pathways (associative mechanism and dissociative mechanism). The following five-step mechanism describes the associative mechanism for the ORR on CoO faces

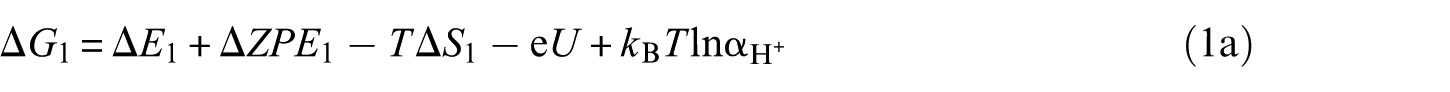

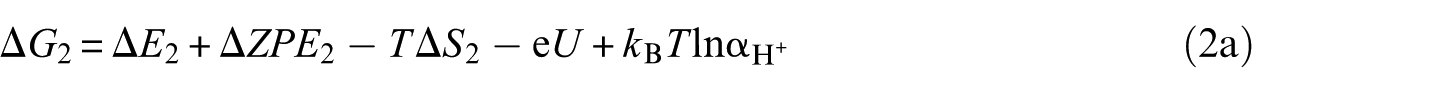

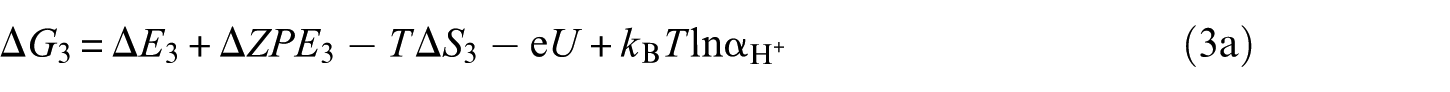

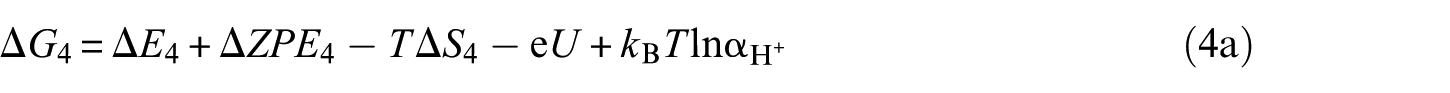

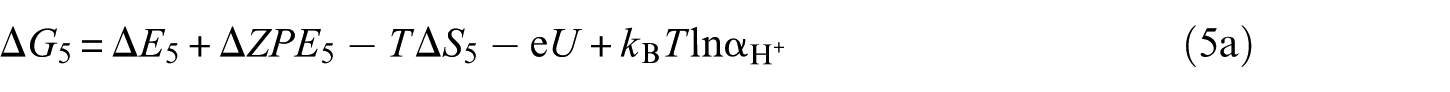
The lone ‘*’ symbol represents the reaction site in the slab models. The symbols ‘O2*’, ‘HOO*’, ‘O*’ and ‘HO*’ represent the surface with corresponding chemisorbed species. The mechanism involves four reduction steps (steps 2–5), each of which results in the transfer of one proton and one electron. O2 first adsorbs onto the reaction active site. The farther oxygen atom on the surface reacts with H+ and e− to form the HOO*. In the next step, a water molecule is released and O* is formed. In the last two steps, the oxygen molecules on the surface are reduced to another water molecule after two proton-coupled electron transfers. The change in Gibbs free energy (ΔG) during the chemical reactions is shown in equations (1a)–(5a). The free energy surfaces presented and discussed in this work are at standard conditions (T = 298.15 K, pH = 0). ΔE is the adsorption energy calculated from DFT. The zero point energy (ZPE) was also calculated by the DFT calculation of the vibrational frequencies. Enthalpy changes with temperature (from 0 to 298 K)43–45 were neglected, such changes being generally expected to be very small. T is temperature (298.15 K), eU is the shift in electron energy when a bias is applied and
The following dissociative mechanism was also taken into our consideration to identify fundamental aspects of ORR. In contrast to the associative mechanism, after O2 adsorbs onto the reaction site, the oxygen molecules form two oxygen atoms adsorbed onto the surface, respectively. One of the oxygen atoms then undergoes two subsequent reduction steps to generate the first water molecule. The last two reduction steps are the same as those in the associative mechanism. Throughout the process, two oxygen atoms produce two water molecules without generating H2O2 as an intermediate 47

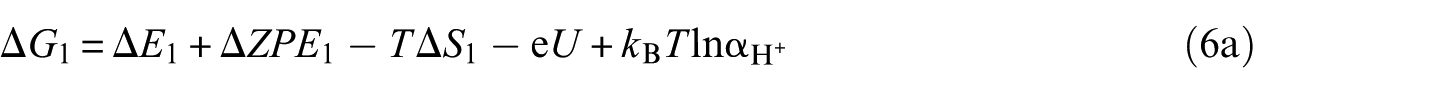

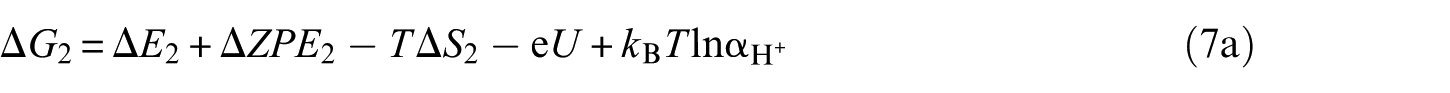

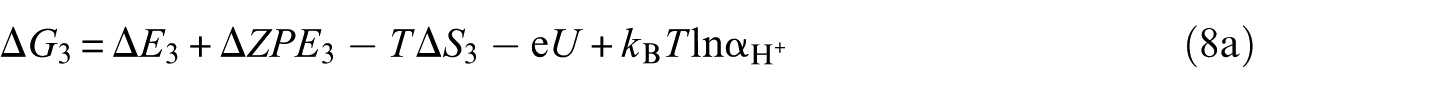

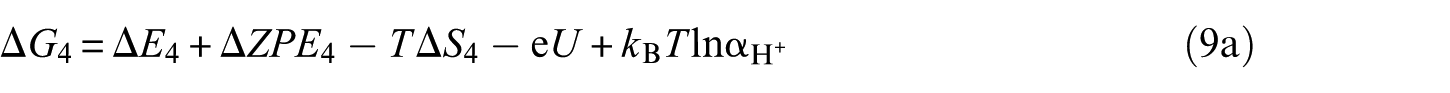

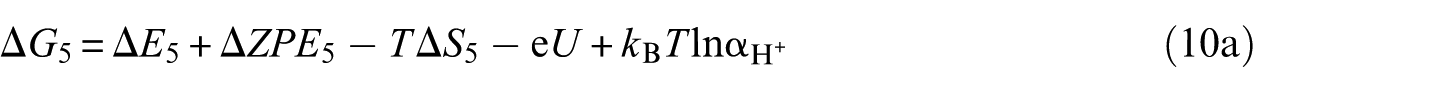
The results are summarized here to compare the ORR process on the CoO (111) and CoO (100) surfaces based on different reaction mechanisms. The optimized structures of the adsorbed species on the two different surfaces are illustrated in Figure 2, numbered sequentially as 1–7 and 8–14. To study the ORR activity of the CoO (111) and CoO (100) surfaces under electrochemical conditions, the free energy diagrams of all ORR intermediates were calculated according to the method developed by Nørskov et al. 16 The free energies of the all elementary steps including ZPE, and entropy corrections are listed in Table 1.
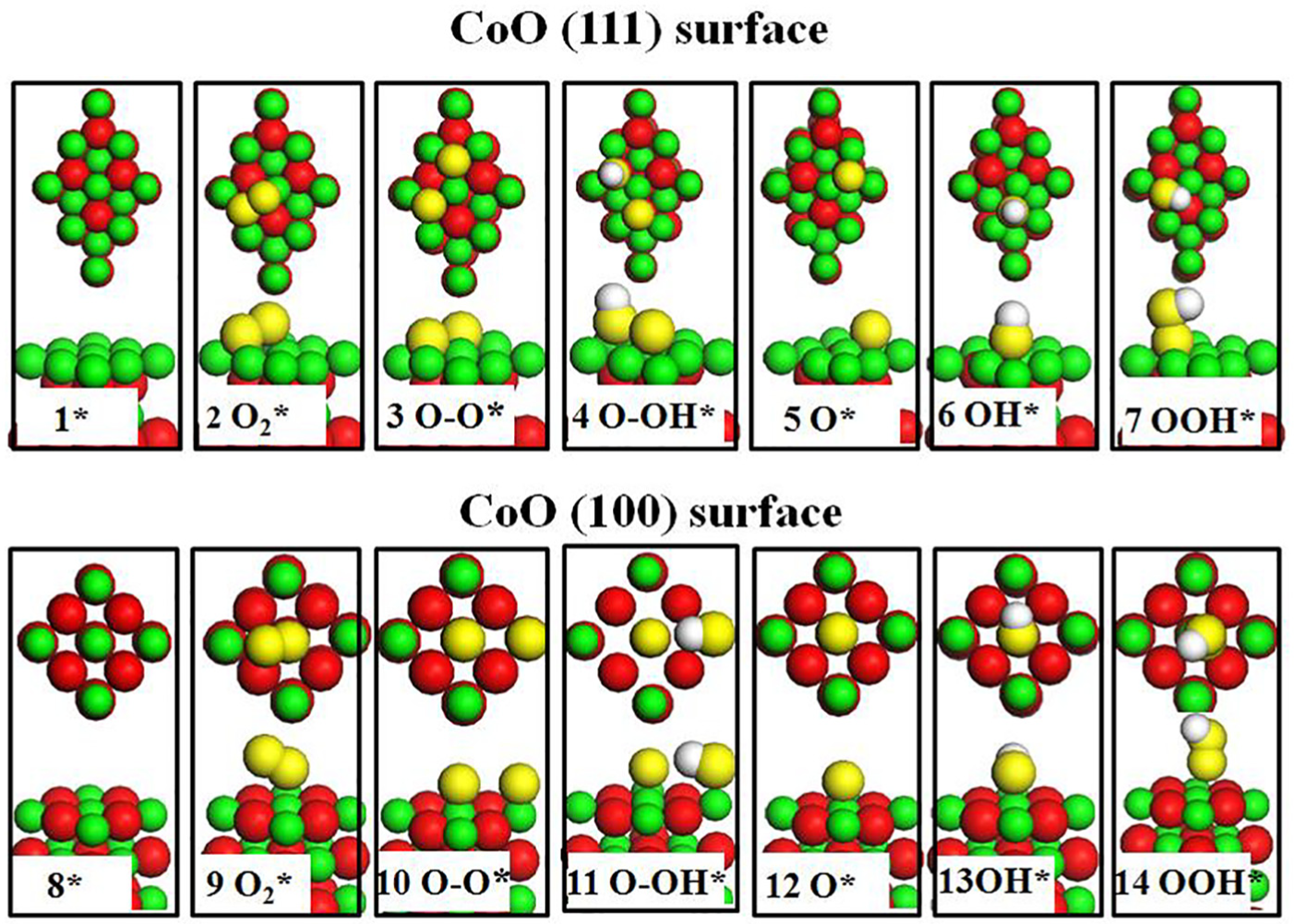
Optimized structures of all the species during ORR, states 1–7 and 8–14 correspond to the CoO (111) surface and CoO (100) surface, respectively. All the optimized structures include top and side views. The oxygen atoms from water and molecular oxygen are coloured in yellow, the remaining O and Co atoms are coloured in red and green, respectively.
Calculated free energy changes in the ORR elementary steps on the CoO (111) and CoO (100) surfaces.
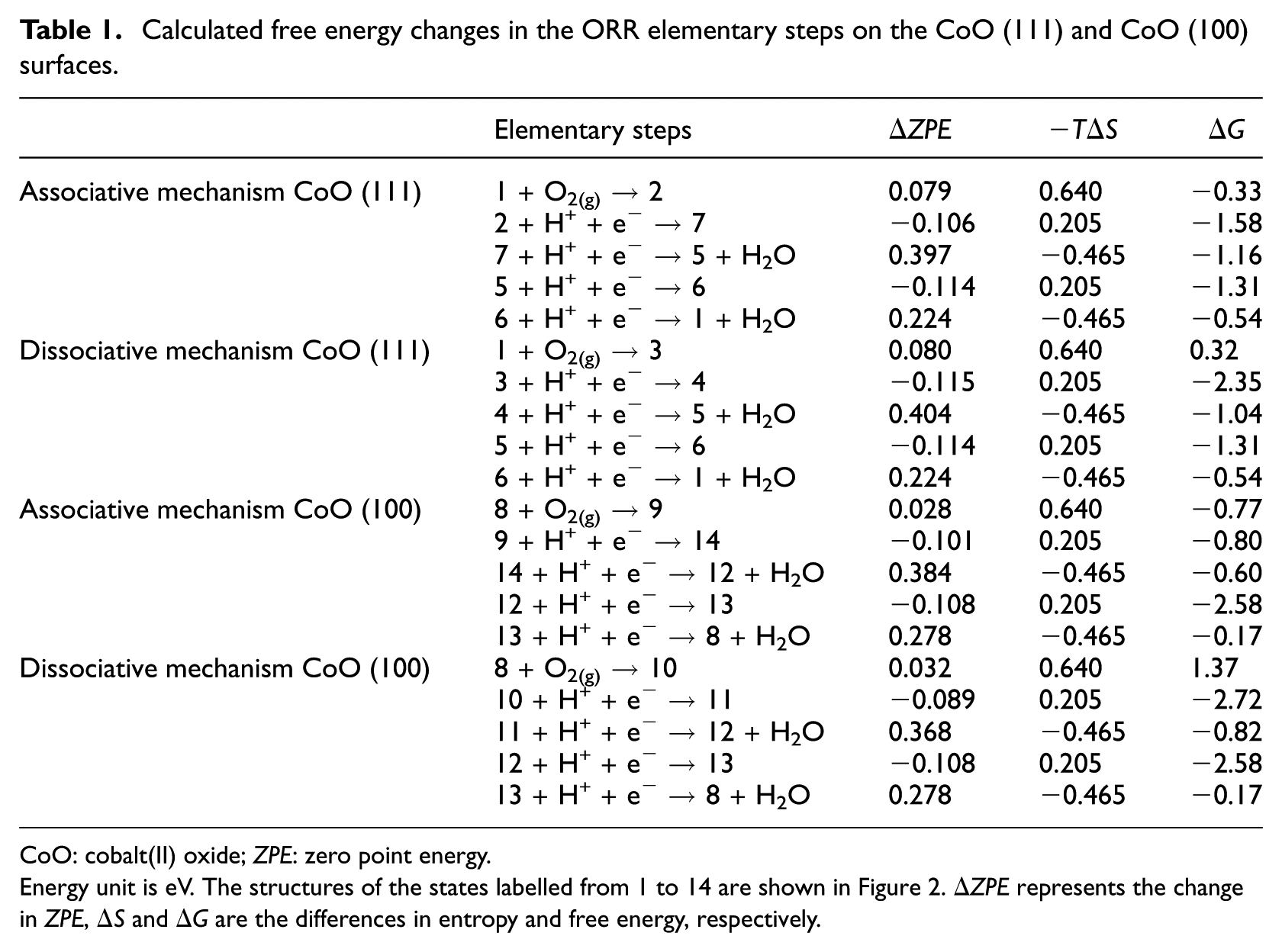
CoO: cobalt(II) oxide; ZPE: zero point energy.
Energy unit is eV. The structures of the states labelled from 1 to 14 are shown in Figure 2. ΔZPE represents the change in ZPE, ΔS and ΔG are the differences in entropy and free energy, respectively.
We have built the ORR free energy diagram for each established model by obtaining the free energies for all reaction steps. The Gibbs free energies of the intermediates are shown in Figure 3. Based on either associative or dissociative mechanisms, the first step in the ORR process on the CoO (111) and CoO (100) surfaces is the O2 adsorption process. In the associative mechanism, O2 adsorbs on the CoO (111)/(100) surface. The overall change in free energy follows the four-electron reduction of O2 to H2O. The free energies of intermediates at different potentials are shown in Figures 3(a) and (c). In the dissociative mechanism, when the oxygen molecules attack the active site of CoO (111)/(100), O2* decomposes rapidly into O-O*. The two co-adsorption oxygen atoms go through two hydrogenation reaction steps respectively to generate two water molecules. The free energies of the intermediates are shown in Figures 3(b) and (d) at the potentials of 0.0 and 1.23 V, respectively.
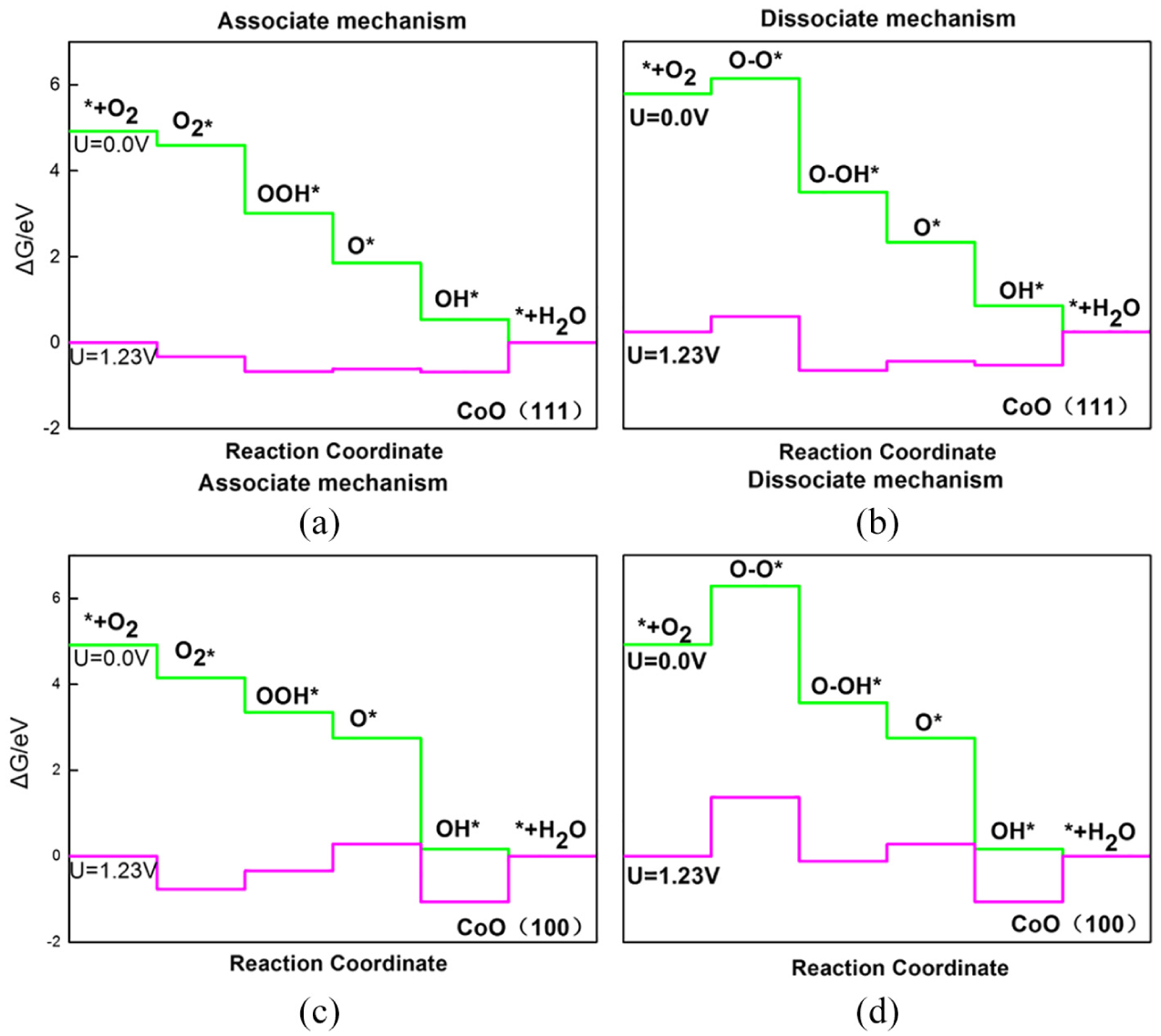
Free energy diagram at pH = 0 and 298 K for the five steps of the ORR at zero potential (V = 0) and equilibrium potential (V = 1.23 V): (a) associative mechanism on CoO (111), (b) dissociative mechanism on CoO (111), (c) associative mechanism on CoO (100) and (d) dissociative mechanism on CoO (100).
It was found that the change in Gibbs free energy of oxygen adsorption on the reaction site is −0.33 eV based on the associative mechanism and 0.32 eV based on the dissociative mechanism on CoO (111). At the same time, the ΔG is also different on CoO (100), being −0.77 and 1.37 eV for the associative and dissociative mechanisms, respectively. Moreover, the change in Gibbs free energy of the first step was also quite different. This value was negative in the associative mechanism and positive in the dissociative mechanism. Therefore, we propose it is more feasible that oxygen is in molecular form when adsorbed on the surface. On the one hand, when the reduction was of OOH* or O-OH* to O* and one water molecule, the change in Gibbs free energy was calculated to be −1.16 eV on CoO (111) while it was −1.04 eV on CoO (100). On the other hand, when the reduction was of OH* to * and another water molecule, the ΔG was −0.54 eV on CoO (111) while ΔG it was −0.17 eV on CoO (100). These results clearly illustrate the better catalytic activity on the surface CoO (111). The free energy diagram in the associative mechanism on CoO (111) is relatively constant, as shown in Figure 3(a). Based on these data for the Gibbs free energy on CoO (111), when O2* reacts with two H+ to generate O*, the ΔG is −1.58 and −1.16 eV, respectively, i.e. they are all close to 1.23 eV. However, the data for the dissociative mechanism are −2.35 and −1.04 eV for each H+, respectively. The results indicate that the first oxygen atom is more likely to react and convert to a water molecule in the associative mechanism.
DFT reveals the energy of each structure as shown in Figure 4. The energy of the CoO (111) structure ranges from −202 to −182 eV, while the energy of CoO (100) ranges from −156 to −138 eV, which is due to the different numbers of atoms in their structures. O2* has a higher energy than O-O*, OOH* and O-OH* regardless of whether the adsorbates are on CoO (111) or CoO (100) or not. This demonstrates that OOH*, O-O* and O-OH* are relatively more stable.
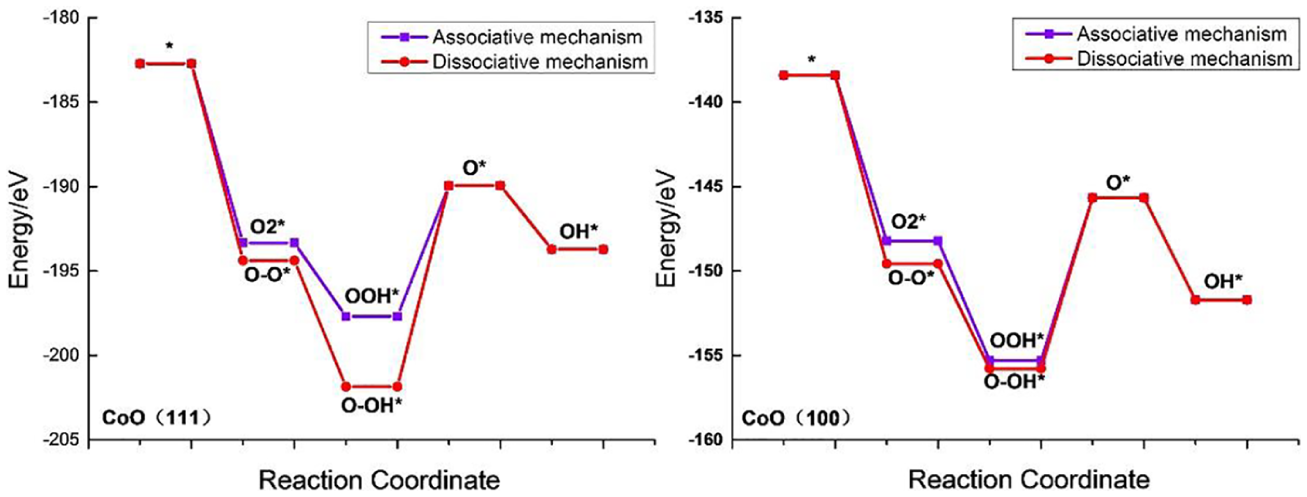
Energy diagram of ORR processes on CoO (111)/CoO (100) in the associative and dissociative mechanisms. The purple line is the associative mechanism and the red line is the dissociative mechanism.
The electronic configurations of the CoO (111)/CoO (100) structures were then analysed using the total density of states (DOS) and the partial density of states (PDOS). The DOS and PDOS for CoO (111) are plotted in Figure 5(a), while the DOS and PDOS for CoO (100) are plotted in Figure 5(b). From Figures 5(a) and (b), it can be observed that the occupied states below the Fermi energy (0 eV) are of mixed Co 3d and O 2p character. Furthermore, the majority of Co 3d and O 2p states have energies ranging from −8 to −0.5 eV. On the other hand, the occupied states of the valence band are mainly caused by Co 3d, which largely contributed below 4 eV. Compared with O 2p, the Co 3d state made a great contribution whether at the top of the valence band or the bottom of the conduction band, illustrating that Co plays an important role in electron transport. A Co 3d sharp peak appears at a valence band of 2.0 eV. This peak suggests the change of 3d in the electronic structure of the system, which plays a major role in the electron transport when catalyzing the ORR.
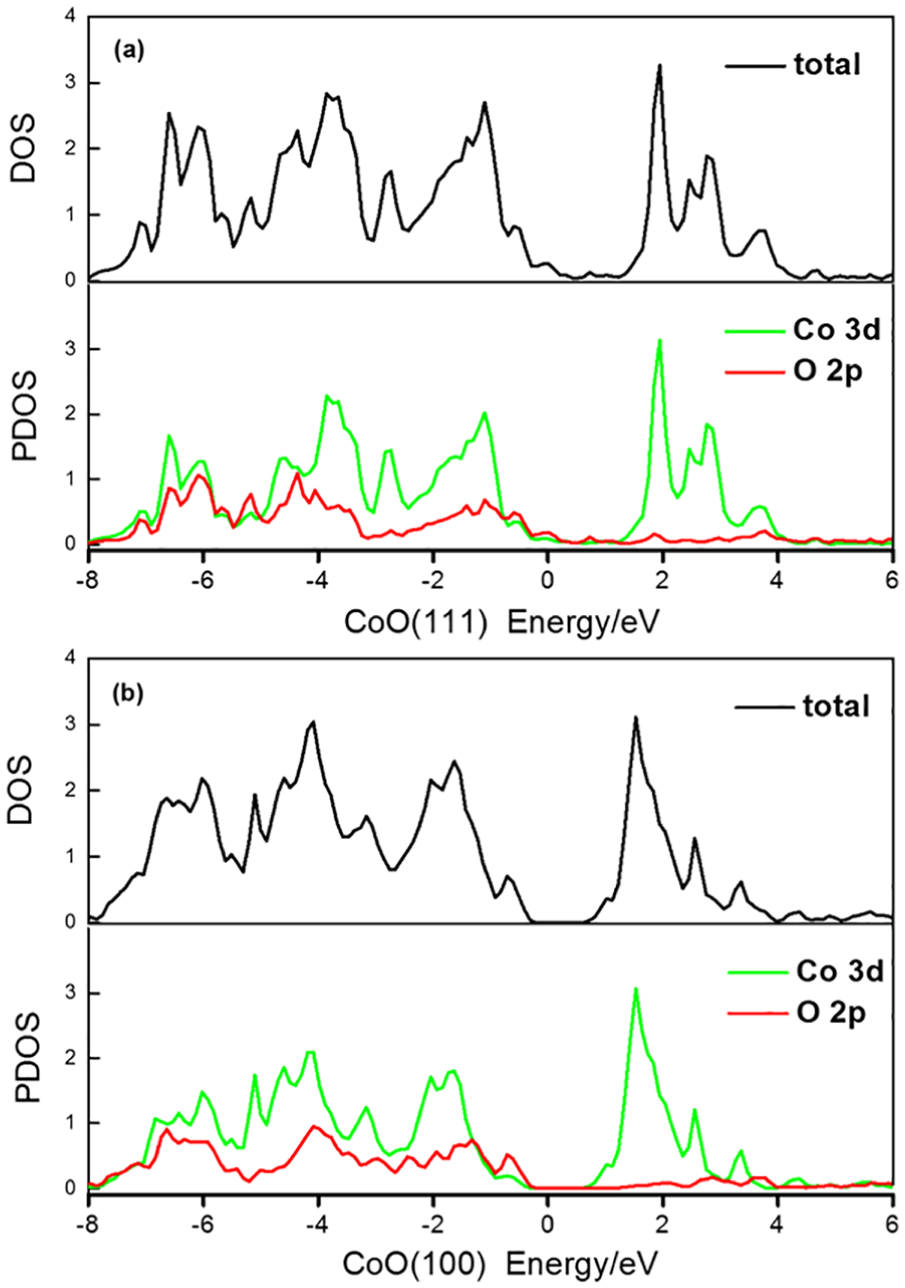
DOS and PDOS of (a) CoO (111) and (b) CoO (100) structures.
In addition, it is noted that the Co 3d centre of CoO (111) is closer to the Fermi energy than that of CoO (100), as shown in Figure 5. The closer the d zone of an atom is to the Fermi energy, the stronger is its adsorption capacity for external molecules. Thus, in the first step of the ORR process, the adsorption capacity of the Co site on CoO (111) is better than that of CoO (100), which should result in the higher ORR intrinsic activity of CoO (111). This agrees well with our energy diagram findings.
Conclusions
In summary, the electrocatalytic ORR mechanism on CoO (111) and (100) surfaces was investigated using DFT calculations. ORR intermediate structures based on both associative and dissociative mechanisms were constructed and optimized. As a result, the ORR Gibbs free energies on the CoO (111) and (100) surfaces based on the associative mechanism were calculated to be smaller than those based on the dissociative mechanism, demonstrating that the associative mechanism should be more likely to be the ORR pathway. Moreover, the electrocatalytic ORR activity on the CoO (111) surface was found to be higher than that on CoO (100). This work should not only provide theoretical understanding of the ORR mechanism on CoO (111) and (100) surfaces but also shed light on future design of highly active CoO ORR catalysts for fuel cells or other next-generation energy devices.
Footnotes
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship and/or publication of this article.
Funding
The author(s) received financial support for the research, authorship, and/or publication of this article: This work was supported by the Natural Science Foundation of China, the Programme for Changjiang Scholars and Innovative Research Team in the University, the Fundamental Research Funds for the Central Universities, the National Key Research and Development Project (grant no.: 2016YFF0204402) and the long-term subsidy mechanism from the Ministry of Finance and the Ministry of Education of PRC.