
Abstract
Keywords
Background
Digital health interventions have the potential to improve the quality of healthcare and have been subjects of extensive research and implementation during the last years. Interventions and solutions range from Mobile Health (mHealth) apps, telemedicine, Artificial Intelligence (AI), virtual reality to electronic patient records, digital biomarkers and sensors. These technologies mainly differ in their intended use and access such as being freely available well-being tools or rigorously regulated medical devices.1–3
Regulatory bodies within the medical affairs sector are dedicated to ensuring public health and patient safety by overseeing the production, use, and monitoring of medical devices. 4 Analogous to drug development, medical devices should not only be safe but also show efficacy. Within this realm, there exists a subset known as Software as a Medical Device (SaMD), with Digital Therapeutics (DTx) falling under this category. They emphasize on software-based solutions over hardware and are subject to medical device regulations. According to the International Medical Device Regulators Forum (IMDRF) “a ‘Software as a Medical Device’ is defined as software intended to be used for one or more medical purposes that perform these purposes without being part of a hardware medical device.” 5 Compared to SaMD, DTx are characterized by a more comprehensive and patient-directed approach, providing a greater level of detail and specificity in the definition. The Digital Therapeutics Alliance defines them as “delivering evidence-based therapeutic interventions to patients that are driven by software to prevent, manage, or treat a medical disorder or disease. They are used independently or in concert with medications, devices, or other therapies to optimize patient care and health outcomes.” 6 To gain market access, both SaMD and DTx must receive approval from regulatory bodies and, in some cases, secure certifications such as the CE (Conformité Européenne).7,8
Digital health technologies, including SaMD and DTx, are employed for collecting data related to digital biomarkers. However, the concept is still emerging, marked by varying or incongruent definitions and the regulatory pathways for digital biomarkers are fragmented.9–11 The Food and Drug Administration (FDA) established a definition for a digital biomarker as being “a characteristic or set of characteristics, collected from digital health technologies, that is measured as an indicator of normal biological processes, pathogenic processes, or responses to an exposure or intervention, including therapeutic interventions.” 9 The FDA and the European Medicines Agency (EMA) propose to use biomarkers to accelerate the development of medical products.12–14
As the regulatory framework often depends on slow and rigid processes, it can happen to lag behind innovation. 15 Existing literature found the complexity and heterogeneity in regulations to be impeding the development of products that are internationally available and competitive. 16 Lack of knowledge in regulatory science was identified to be a major barrier for the commercialization despite numerous inventions in academic research. 17
Therefore, we aimed to provide an update on the most relevant regulations required to gain market access by obtaining approval from the regulatory bodies. We analyzed regulations, guidelines, policy papers, peer-reviewed papers, industry documents and government websites from the United States (US) and the European Union (EU) to identify the most relevant aspects, rules and standards necessary to test and validate digital health solutions in the field of digital biomarkers and DTx. The review will begin by discussing relevant legal frameworks for medical devices, with a particular focus on SaMD in the US and the EU. This includes the process of risk classification, premarket submission, applicable software standards, technical documentation, and clinical evaluation requirements. Next, the regulations specific to DTx as a subset of SaMD will be examined in greater detail. The subsequent chapters will cover (digital) biomarker qualification and the role of digital elements in clinical studies. Finally, the review will address applicable data protection laws and other ethical considerations.
Methods
This narrative review synthesizes the regulatory requirements for developing and bringing DTx and SaMD to market in the US and the EU. The review aimed not only to identify similarities and differences in regulatory approval pathways but also to explore emerging trends related to reimbursement models for DTx, the integration of AI, and the use of digital biomarkers and other digital health technologies in product development.
An unstructured literature review was performed, incorporating regulatory documents, policy papers, peer-reviewed publications, industry reports, and official websites of government and regulatory authorities. The search was guided by keywords such as SaMD, DTx, digital biomarkers, and CE in the context of the FDA, EMA, and IMDRF frameworks.
Roadmap of regulatory requirements for digital therapeutics
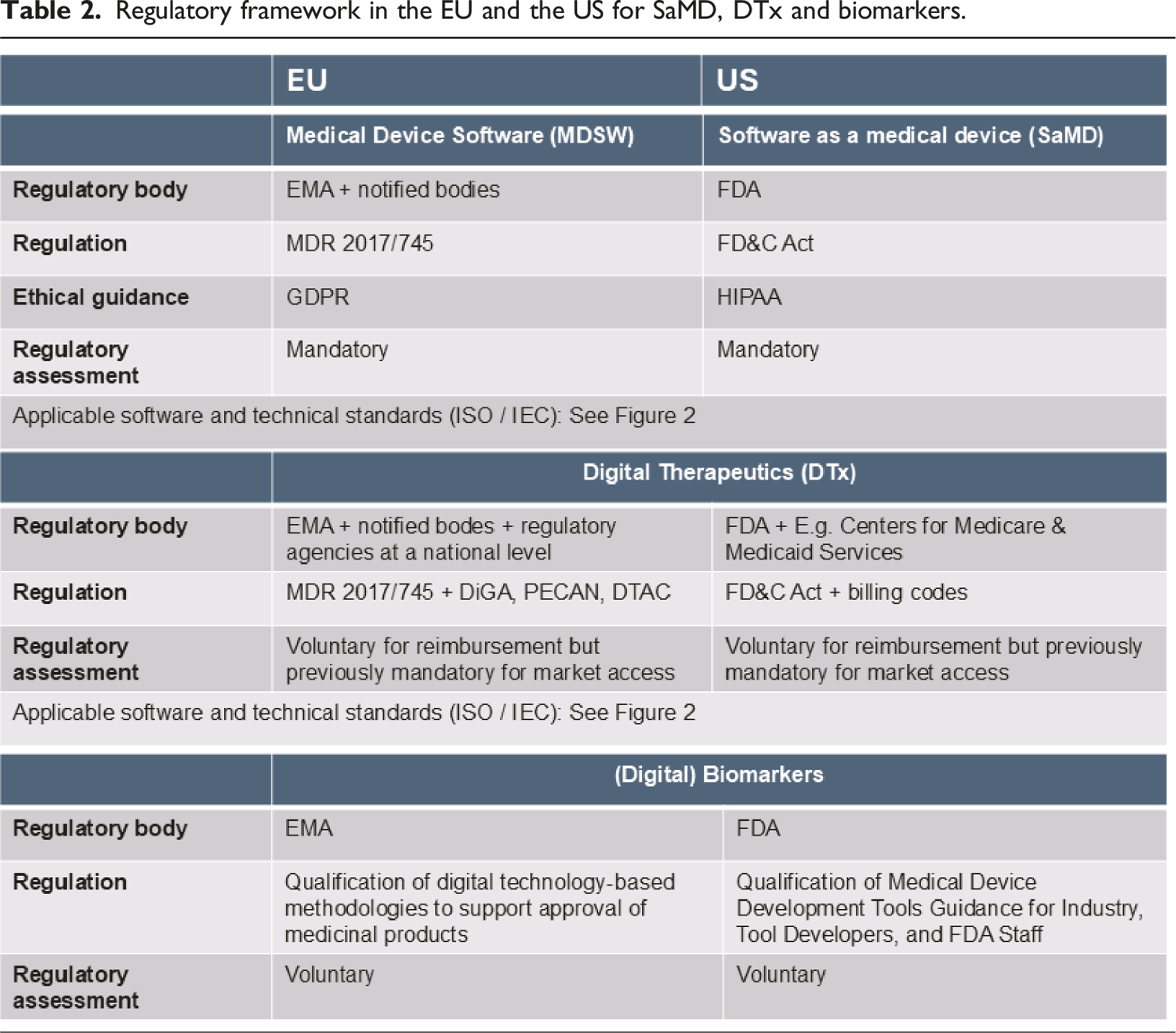
Developers of DTx and SaMD need to identify the applicable regulations. Depending on the geographical context the relevant regulatory bodies are either the US FDA and its Center for Devices and Radiological Health (CDRH) or the EMA.18,19 On the international level, the relevant body is the IMDRF which aims to harmonize the regulatory framework for medical devices internationally. 20 The EMA and the FDA key regulations on medical devices are the MDR 2017/745 (EU) and the Code of Federal Regulations (CFR) Title 21 with the Food, Drug and Cosmetic Act (FD&C Act), subcategory H, respectively.21–23 Additionally, on the FDA website, the most important guideline documents for SaMD can be found under the Medical Devices section.
In May 2021, the European Medical Device Regulation (MDR) came into force and replaced the Medical Device Directive (MDD) 93/42/EEC. With its new regulation, the EU aims to increase safety for patients through the application of stricter measures. Relevant adjustments in the new regulation involve reclassification of some devices, increased scope, provision of clinical evidence through a clinical study and premarket expert-panel consultation for high risk devices, elevated post-market surveillance and studies including reports, unique device identifier and stricter requirements for claiming equivalence. 24 On the 15th of March 2023, the European parliament and council approved an extension of the transition period from the MDD to the MDR that should have ended in May 2024. The European commission proposed an extension until December 2027 and December 2028, depending on the risk classification of the medical device, to avoid shortages in supply. 25
To prepare a submission for market approval either by the FDA or the EMA’s assigned notified bodies, manufacturers have to identify in a first step whether their software classifies as a SaMD. Several guidance documents were issued to facilitate the process for developers. As a rule of thumb, software that do not influence the decision making in health related situations of medical personnel or users, do not classify as a SaMD (e.g. administrative support or general wellbeing apps).26,27 In the case manufacturers are developing a DTx, it will always fall under the category of a SaMD.
Under the CDRH, biomarkers are classified as Medical Device Development Tool (MDDT) and can be qualified by a voluntary submission to the FDA. 13 A key document for biomarkers in the US is the ‘Qualification of Medical Device Development Tools Guidance for Industry, Tool Developers, and FDA Staff’. However, there are no specific guidelines for digital biomarkers. The EMA regulates biomarkers with the ‘qualification of digital technology-based methodologies to support approval of medicinal products’ document but also on a voluntary basis. 28
Risk class evaluation
To obtain a medical device certification, for example the CE label and subsequently access to the market, producers are required to identify the risk class of their software.29,30 Risk classes range from class one, considered for minimal risk devices, to class three which represent high risk devices.21,29 Guidance to choose the correct category is given by the MDR, the CFR and the IMDRF, as the classification in the US and Europe is similar, but not exactly the same.5,21,22,27,29 Figure 1 illustrates the slightly different categorizations according to the FDA and the EMA. Additionally, the EU’s special working group Medical Device Coordination Group (MDCG), released a guidance document to help the industry correctly classifying their devices.
31

Premarket submission
In the US, once the appropriate risk class has been determined, the selection of the corresponding type of premarket submission follows. Premarket submission includes design controls to ensure quality assurance, nonclinical testing, clinical evidence, consensus standards such as International Organization for Standardization (ISO) compliance and labelling. The type of premarket submission also depends on whether a similar device already exists and assurance of safety and effectiveness of the device has to be included in the premarket submission. Premarket submission types include investigational device exemption, premarket notification (510(k)), premarket approval application, de novo or humanitarian device exemption. Other requirements from the FDA are the compliance with Good Manufacturing Practices (GMP) and listing the device.23,32,33
In the EU, conformity assessment is executed by notified bodies of member states. In some cases, expert panels who work together with the EMA can or have to be consulted. Technical documentation, safety and performance of the device and the quality system of manufacturers are reviewed. Most important for manufacturers are the General Safety and Performance Requirements (GSPR) in the MDR Annex. The provision of a risk management, clinical evaluation, technical documentation, quality management, a post-surveillance system and device identification and registration are key elements of the GSPR.7,21,34 In order to keep the CE conformity label, regular recertifications by these notified bodies is necessary. 30
Although the US and the EU have separate market approval pathways, they both require a unique device identifier to be placed on the medical device and a corresponding database. Standardization efforts and collaboration with the IMDRF to allow communication between these databases and recognition among the global market are aspired by the US and the EU. 35
Software standards and technical documentation


Clinical evaluation of software as a medical device
Besides the technical documentation, the MDR and FDA require clinical- and performance evaluations. Manufacturers should take into account ISO 14155 “Clinical investigation of medical devices for human subjects — Good clinical practice”, GMP and the special guidelines for clinical evaluation and performance published by the MDCG and the CDRH.21,47–51 The use and application of these international standards in the US and the EU facilitate the acceptance of clinical trial results obtained outside of the concerned geographical area. 50
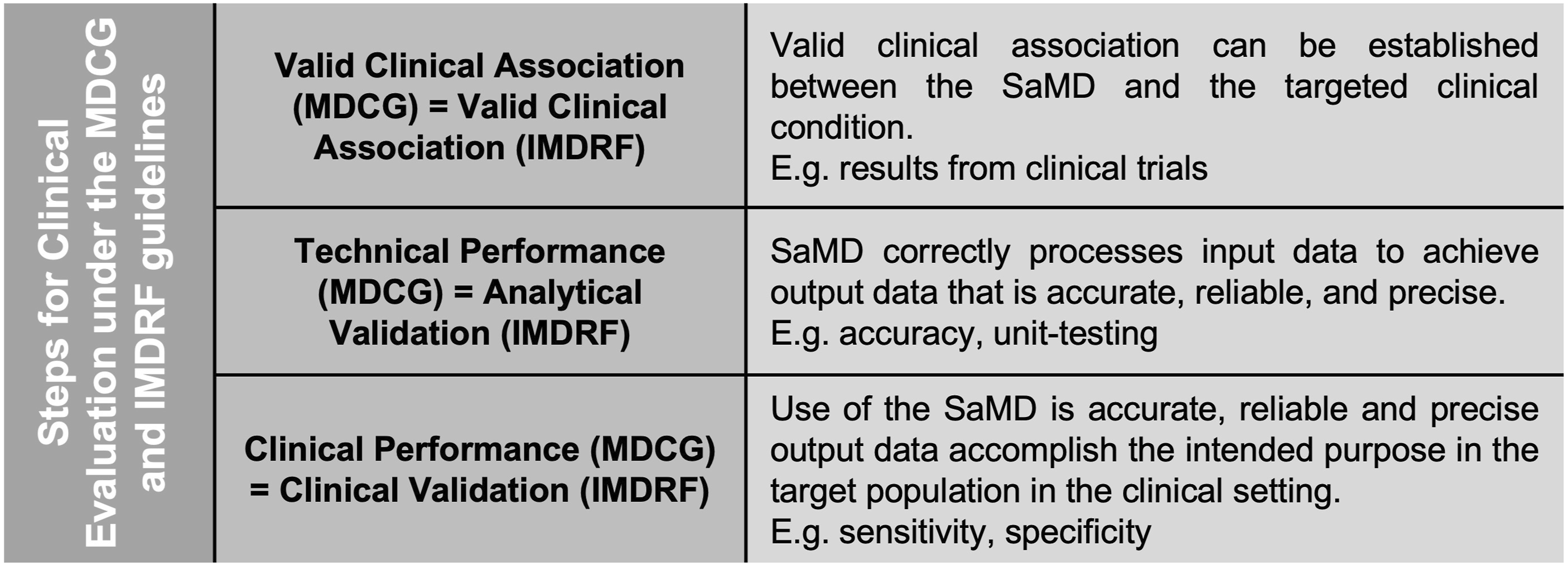
Under the MDR and the MDCG guidelines, clinical evaluation includes the demonstration of valid clinical association, technical performance and clinical performance. The clinical evaluation aims to show the clinical benefit of a SaMD, as claimed by the manufacturer, and is dependent on its quality and amount of supporting data.
A valid clinical association is either demonstrated, or can be already existing evidence and highlights a connection between the generated output and the targeted condition. Acceptable evidence can either originate from clinical trials, proof of concept studies, literature or professional guidelines.
The validation of technical performance and analytical capability entails demonstrating the SaMD’s precision, accuracy, and reliability in producing the intended output based on the provided input data. Verification and validation activities, such as unit-level, integration and system testing, as well as the utilization of curated databases, curated registries, reference databases, or previously collected patient data, can contribute to the generation of evidence supporting the technical and analytical performance of a system. 47
To provide clinical performance, a SaMD has to create an output that has a positive and relevant impact on a patient’s health, diagnostic, monitoring or management and be in line with the stated purpose. Measures for clinical performance are for example sensitivity, specificity, positive predictive value, negative predictive value, number needed to treat or odds ratios. 47 Although manufacturers can sometimes use data from previous equivalence studies, the MDR requires high risk devices to demonstrate safety and evidence in a clinical study. 24
The FDA orients itself on the IMDRF guidance for clinical SaMD evaluation. Their guidance document includes the same requirements, but with slightly different terminology than in the MDCG guidelines as illustrated in Figure 2.
48
Manufacturers should document these steps in a clinical evaluation report. 47 The clinical evaluation is a continuous life cycle for SaMD, which is ongoing after the market approval in form of post-market studies. According to the IMDRF, this can be done by collecting real-world performance data such as adverse events or user feedback, and be used to improve the evidence and functionality for the SaMD. Furthermore, by continuously reevaluating the device, a new allocation of a different risk category could become necessary.24,48
When regulators are evaluating software, it is important to note that they test the functionality of the software but not the mobile platform on which the software is run. Mobile platforms as defined by the FDA are for example smartphones, computer or laptops. This is of high importance as a software run on a smartphone instead of a computer could affect for example displayed image quality. 27 Therefore, it is important for the manufacturer to either specify the platform the software should be run on or show equality between platforms in the clinical evaluation. The FDA evaluation only applies if the software is a medical mobile application which falls under medical device regulation. Here again, the key aspect for whether it is medical mobile app or not is the intended use as previously described. 27
Regulations of digital therapeutics
The terminology for digital health software solutions varies across different countries. In the United States, the predominant term is ‘Software as a Medical Device’ (SaMD), while in Europe, ‘Medical Device Software’ (MDSW) is more commonly used. In Germany, in addition to MDSW, the term DTx is frequently employed. Depending on the country, DTx are fully regulated under the medical device and SaMD framework or have additional regulatory pathways. In the US and the EU, DTx are covered by Chapter 21 of the CFR and the MDR. Contrasting to the MDR, functioning as an umbrella to harmonize the medical device market access for countries in the EU, the approval and reimbursement for DTx is country specific. The main difference is whether and by whom they are reimbursed. For example, Germany has a specialized program regulated by the Bundesinstitut für Arzneimittel und Medizinprodukte, allowing for the prescription of mHealth apps, known as DiGA, which are covered by health insurance. In contrast, other countries lack adapted pathways, relying either on private insurers for reimbursement or individual funding decisions. 52
Evaluation of evidence for digital therapeutics
In cases where manufacturers would like to register their SaMD as a DTx (e.g., a DiGA in Germany), additional evaluations are required for DiGA, which are typically low-risk class devices falling in category 1 or 2a. In Germany, developers have to show interoperability and a positive health care effect which includes direct medical effects on a patient or structure and process improvements. This positive effect can be shown by improving quality of life, adherence, health competency, coordination of care, shorten disease duration, or extending life expectancy and so on. Evidence from conformity assessment can be used as a support but studies for DTx approval have to be conducted in Germany to ensure applicability to the German population. Exceptions are made if there is proof that the context where the study was conducted is highly similar to the German health care environment and German population. Moreover, the regulatory body only accepts quantitative comparable study designs while expert opinions or descriptive studies are rejected. 53 Other evidence to be delivered when manufacturers seek reimbursement concerns the economic aspect of the technology. Achieving reimbursement often requires undergoing a health technology assessment (HTA) on a national level, evaluating not only clinical effectiveness but also cost-effectiveness. 54 From a payer perspective, HTAs are essential to ensure that funding decisions are evidence-based and helps to prioritize interventions that provide meaningful patient benefit while ensuring efficient allocation of limited healthcare resources. 55 In Europe, only Germany with the DiGA, France with the Prise en Charge Anticipée Numérique (PECAN), and the United Kingdom (UK) with the Digital Technology Assessment Criteria (DTAC) have established such reimbursement pathways. In France and the UK, certain cases may additionally require a budget impact analysis or a cost-effectiveness analysis. 54 Notably, the UK has developed a framework for HTA that is specifically tailored to DTx. 55 In the US, from a payer perspective, clinical effectiveness, cost-effectiveness, and the availability of applicable billing codes would be required to reimburse DTx. 56
Biomarker qualification
To obtain the qualification of a biomarker, a reliable way to measure it has to be provided, including test validity measurements. Additionally, strength of evidence and performance characteristics will be examined by the regulatory body. Regulators will look at the evidence for the biomarker independently of the measurement, requiring information regarding the category, context of use, the added benefit and data supporting the relationship between the biomarker and clinical outcome of interest. But without at least one accurate assay the final qualification of a biomarker could fail. Although qualification is voluntary, the process supports standardization and scientific reliability. Biomarker (tests) qualified as MDDTs can help to demonstrate clinical evidence. The use of qualified biomarkers in device development or regulatory submissions can increase safety and facilitate decisions for SaMD approval by the regulatory body.13,28,57
Traditional biomarkers such as blood pressure or heart rate can be classified in various categories, including risk, prognostic, monitoring, safety, diagnostic, predictive or pharmaco-dynamic response biomarker.58,59 The new category of digital biomarkers do not solely consist of data collected through digital health technologies but are also converted into a specific metric, often utilizing an algorithm.9,58 In many cases, this allows for the continuous collection of health-related data over an extended period, rather than just at specific timepoints. This feature is particularly advantageous for monitoring chronic diseases. 59 Digital biomarkers are often cheaper and more convenient for the patients as they are less invasive compared to traditional biomarkers. 59 Transition biomarkers are usually biological indicators that signify a change in disease state or response to treatment.60,61 They are more focused on specific biochemical changes whereas digital biomarkers rely on technology for continuous data collection measuring disease states on a broader range. On the other hand, digital biomarkers may be used to predict the transition from active disease towards remission, thus overlapping with a transition biomarker. An illustrative example of an emerging digital biomarker involves the utilization of AI for the recognition of dorsal finger fold changes, serving as an indicator for monitoring joint swelling in rheumatoid arthritis. 62 Other examples are a potential digital speech and voice biomarker to monitor disease progression in patients suffering from multiple sclerosis 63 or a digital gait biomarker that analyses step variability and body sway to track changes in spinocerebellar ataxia type 3. 64 Another study showed promising results using smartphone-based eye tracking to detect the onset and development of mental fatigue in a time saving manner compared to traditional fatigue measurements. 65 Because of the increased volume of data that can be gathered by digital health technologies, digital biomarkers have significant potential for personalized and precision medicine. 59 There are also cases where parts of a digital biomarker are classified as SaMD. 58 However, regulatory pathways are not yet fully developed for digital biomarkers. Digital biomarker development and their qualification is cumbersome because validation methods and regulatory guidelines are lacking and building the bridge between data collected by wearables or mHealth apps to a validated digital biomarker needs a lot of resources. Moreover, digitally collected health data is subject to data protection laws and people are often hesitant to share this kind of data due to their fear of data security breaches. 66
When digital biomarkers are used to demonstrate clinical evidence, they are often employed as endpoints in clinical studies. Digital biomarkers happen to be especially suitable for remote monitoring of patients as the data can be collected by devices and wearables and are therefore more and more deployed in decentralized or digital clinical trials. 58
Digital biomarkers have a great potential to improve the healthcare system as they serve in different contexts, are mostly non-invasive, can collect data continuously, remotely and passively in big amounts.9,58,59 However, to enable the development of digital biomarkers, guidelines and regulations should be advanced. Therefore, we propose that regulations for digital biomarkers should be integrated in the existing biomarker qualification to prevent additional complexity. Digital biomarkers could be introduced as an eighth category, or each of the seven existing categories could encompass both digital and non-digital biomarkers, e.g. digital monitoring biomarker versus monitoring biomarker. Given that digital biomarkers utilize technologies like software and platforms, there should be an automated verification process to determine if they fall under the medical device regulations or ISO standards as described above. A summary of the roadmap to (digital) biomarker qualification in the EU and the US is illustrated in Figure 3.
The regular integration of AI and algorithms in digital biomarkers warrants specific regulations. That could for example be compulsory transparency regarding the training sets or open-access codes.58,66 Additionally, it is highly recommended that the integration of the FAIR principles for data management should be mandatory when qualifying a digital biomarker. 66 To fully realize the potential of digital biomarkers, it is essential to guarantee interoperability of the used technologies. 58 On an ethical note, since these sensors collect sensitive health data, submitters should provide a detailed outline of how data will be protected and how they will comply with relevant data protection laws when qualifying a digital biomarker. In 2021, regulatory bodies of the US, Canada and the UK defined 10 principles to guide the development of Good Machine Learning Practices (GLMP) to address the increasing use of AI/Machine Learning (ML) in SaMD. 68 As part of an action plan, the FDA outlined measures to advance oversight of AI/ML-based SaMD. These include updating the proposed regulatory framework through draft guidance on Predetermined Change Control Plans, promoting harmonized GMLP via collaborative standards efforts, and strengthening patient-centered approaches fostering transparency and trust in AI/ML technologies. The FDA also highlighted the need for regulatory science initiatives to improve methods for evaluating algorithm robustness, resilience, and bias, as well as advancing real-world performance pilots to guide evidence generation for AI/ML-based SaMD. 69 Building on this Action Plan, the FDA has issued several documents addressing these priorities, including its most recent draft guidance, “Artificial Intelligence-Enabled Device Software Functions: Lifecycle Management and Marketing Submission Recommendations.” 70 The IMDRF has formed a working group for AI/ML-enabled medical devices, issued a technical document for terms and definitions and is aiming to harmonize the GMLP internationally. 71 In the EU, the most relevant regulation is the AI Act, which classifies SaMD that are AI systems, or that incorporate an AI system as a safety component, as high-risk medical devices under the MDR. 72
Regulatory framework in the EU and the US for SaMD, DTx and biomarkers.

Digital elements in clinical studies
Technologies and methodologies applied in clinical research and sources for real-world data include electronic case report forms, wearables, electronic patient reported outcomes, electronic consent, mHealth, AI, remote, virtual and decentralized site visits (decentralized clinical trial elements) or digital biomarkers and are widely used in the approval of medicine products and medical devices.14,73–76
Studies using digital health elements and real-world evidence bring along standard regulatory aspects like the Good Clinical Practice (GCP) or the GMP. 77 Simultaneously, the increasing prevalence of decentralized clinical trials and real-world evidence studies give rise to new regulatory considerations, stemming from the associated challenges and the persistence of these emerging methodologies.53,58
The FDA issued guidelines for the “Use of Real-World Evidence to Support Regulatory Decision-Making for Medical Devices.” 73 Many of the included elements are also applied in decentralized clinical trials and real-world evidence is used to support biomarker qualifications and post-market surveillance of medical devices. 73
Furthermore, the EMA issued guidelines on computerized systems and electronic data in clinical trials. Adherence to these “Guideline on computerised systems and electronic data in clinical trials’ EMA/226170/20212” and fulfilling the standards to collect and process reliable data is especially important in decentralized clinical trials as digitalized systems build a pivotal element of these trials.74,78
Vayena et al. 79 identified seven different countries and one region that released guidance documents for decentralized clinical trials through the appropriate regulatory bodies between 2020 and 2023.
Among those, the US published in response to the COVID-19 pandemic a guidance document for the conduct of clinical trials during the public health emergency. The main goal of the non-binding recommendations is to help clinical trial staff to adhere to the GCP, enable the continuation of the clinical trials while guaranteeing participants safety. It halts sponsors to assess alternative options beside normal site visits as documented in the protocol. Close collaboration with ethic committees for protocol changes and implementation of decentralized clinical trial elements is recommended. 80
The EU issued a recommendation paper regarding decentralized clinical trials, primarily as a reinforcement of this emerging methodology that demonstrated success during the pandemic. Similar as the recommendations of the US, the European Commission stresses respecting the safety, dignity and rights of participants when using decentralized clinical trial elements. Secondly, data reliability is key, especially when it’s used for regulatory submissions or publications. 78
Identifying the correct regulations that apply is a challenge in decentralized clinical trials in relation to medical devices. If the medical device already is approved by the regulatory body it has to be marked as such, the use of it has to be compliant with the intended use and the trial will not fall under the medical device regulation. However, in case it is not yet approved the clinical trial will be classified as a clinical investigation of a medical device. Clinical trials can have a mixed format and therefore, regulations can be intertwining. This is not only challenging for the investigators but also the ethical review boards.79,81
Data protection and other ethical aspects
DTx and digitalized clinical trials are newer concepts and their use in studies require the approval by ethic committees. Principal ethical document under the MDR is the World Medical Association Declaration of Helsinki on Ethical Principles for Medical Research Involving Human Subjects.21,47,78,80
While privacy and confidentiality of data are the main ethical issues related to DTx, insufficient digital health literacy, unequal access to technology and possible addiction were other ethical concerns identified in the literature. 82 Ethics reviewers showed hesitancy towards decentralized clinical trials with concerns about the digital literacy, safety of participants and the integrity of the research. 83
The most relevant regulations in terms of data protection are the General Data Protection Requirements (GDPR) in the EU and the Health Insurance Portability and Accountability Act (HIPAA) in the US.84,85 They regulate data privacy and security and are of special importance for digital health, as it affects sensitive data handling. 86 GDPR has to be respected, even outside of the EU if it concerns EU residents and their data. Violations are sanctioned by compensation fees. 85 To ensure compliance with GDPR, developers of SaMD have to respect requirements on a technical, organizational and physical level. 86 Digital biomarker tests, DTx, SaMD and therefore medical device manufacturers often collect passive and raw data of patients. While GDPR easily grants patients access to these data, patients in the US have comparably less possibility to access their digital health data as the HIPAA is less strict. 87 The updated version of the HIPAA does still not cover health data collected by consumer wearables and better regulation from the government and regulatory bodies is demanded. 88 Although non-binding, the FDA recommends in their guidelines to share patient’s data from medical devices upon request.89,90 Besides national data protection laws, the IMDRF and MDCG published guidelines on cybersecurity that should be followed including recommendations for the employment of different software standards.91–93 In the US, section 524B “Ensuring Cybersecurity of Devices”, was added to the FD&C Act. 94
Limitation and outlook
This review only considers regulations from the US and the EU. Future research should encompass the analysis and comparison of regulations from other IMDRF-affiliated countries, including, for example, Brazil and China. Additionally, it should consider countries and their regulatory frameworks unrepresented in the IMDRF, particularly those from the southern hemisphere, to include different perspectives and insights and capture new developments in regulatory science. A list of affiliated members, official observers and countries in the management committee of the IMDRF can be found in the Supplemental Material 1.
Recommendations
While voluntary qualification of biomarker tests by regulatory bodies exists, it lacks specific guidance for various types, including digital biomarkers. The qualification of biomarkers should include a category for digital biomarkers to address gaps in existing regulations and prevent adding complexity with entirely new regulations. This extension should encompass the specifics of the digital aspect, such as data protection, ISO standards, and software that may fall under the MDR. So far, some countries issued position papers and recommendations on digital health elements in studies, but clear regulatory pathways are lacking. Different formats of decentralized clinical trials using medical devices or investigating devices with overlapping regulations has been identified as complexity for manufacturers and ethical reviewers. The clinical trials transformation initiative, a public-private partnership between the FDA and the Duke University, in collaboration with international stakeholders, sets an attempt to establish homogenous and internationally valid rules. 76
Conclusions
While the IMDRF aims to harmonize regulatory requirements, developers of SaMD must still navigate distinct processes when seeking access to multiple global markets, which is very resource-intensive in terms of finances and time. 16 On top of that, if the SaMD is a DTx, obtaining the CE label does not guarantee automatic reimbursement by health insurances. Whether and how they are getting reimbursed is depending on national rules. 52 Regulatory pathways beyond the MDR or Chapter 21 CFR such as the DiGA in Germany are rare but could serve as an example to be followed by other countries.
Enhanced versions of SaMD in form of mobile apps also called DTx bring along concerns about data safety and privacy. Although, they have their justification, they are being addressed through the coverage of the existing regulatory framework of SaMD. Notably, the amendment of the MDR requiring post-market surveillance and re-evaluation of the risk category provides high data privacy and safety for participants. However, protection of sensitive health data is a concern if they are collected by wearables that do not fall under the SaMD/DTx framework. Currently, this type of data is not safeguarded by the HIPAA.
With the emergence of different formats to validate and support SaMD approval, the international community gets a new chance to develop guidelines and processes applicable beyond national borders. Regulations for DTx, decentralized clinical trials and digital biomarkers are converging as manufacturers are increasingly leveraging these digital health technologies in conjunction to develop and evaluate SaMD and DTx.
Supplemental Material
Supplemental Material - Navigating through regulatory frameworks for digital therapeutics and biomarkers
Supplemental Material for Navigating through regulatory frameworks for digital therapeutics and biomarkers by Cinja Koller, Marc Blanchard, Thomas Hügle in Health Informatics Journal
Footnotes
Author contributions
All authors meet the journal’s authorship guidelines. CK and TH contributed to the design and conception of the work. CK has drafted the manuscript. TH and MB substantively revised the manuscript. All authors read and approved the final manuscript.
Funding
The authors received no financial support for the research, authorship, and/or publication of this article.
Declaration of conflicting interest
The authors declared the following potential conflicts of interest with respect to the research, authorship, and/or publication of this article: MB and TH are shareholders of Atreon Société Anonyme. TH is patentholder of Detectra and scientific advisor of Vtuls.
Data Availability Statement
All relevant data can be found in the manuscript.
Supplemental Material
Supplemental material for this article is available online.
Appendix
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.