
Abstract
Due to the huge costs associated with new drug discovery and development, drug repurposing has become an important complement to the traditional de novo approach. With the increasing number of public databases and the rapid development of analytical methodologies, computational approaches have gained great momentum in the field of drug repurposing. In this study, we introduce an approach to knowledge-driven drug repurposing based on a comprehensive drug knowledge graph. We design and develop a drug knowledge graph by systematically integrating multiple drug knowledge bases. We describe path- and embedding-based data representation methods of transforming information in the drug knowledge graph into valuable inputs to allow machine learning models to predict drug repurposing candidates. The evaluation demonstrates that the knowledge-driven approach can produce high predictive results for known diabetes mellitus treatments by only using treatment information on other diseases. In addition, this approach supports exploratory investigation through the review of meta paths that connect drugs with diseases. This knowledge-driven approach is an effective drug repurposing strategy supporting large-scale prediction and the investigation of case studies.
Introduction
Drug repurposing, which is the process of finding new indications for existing drugs, has demonstrated advantages over de novo drug design, 1 including faster development time and reduced risk. 2 It is estimated that drug repurposing can reduce the 10- to 17-year drug development process to 3 to 12 years. 3 Reportedly, drug repurposing constitutes 10 to 50 percent of a pharmaceutical company’s R&D expenditure. 4 Owing to the advantage of increased productivity, drug repurposing has been extensively studied under different names such as drug repositioning, reprofiling, redirecting, and rediscovery. 5 Despite great interest from both academia and industry, the number of successful repositioning cases is relatively small, with most identified through serendipitous observations.6–8 Recently, due to the increased number of public databases and advanced informatics methods, computational or in silico approaches to drug repurposing have attracted great attention. Compared with experimental approaches that utilize actual drugs for screening, computational approaches are both time-efficient and cost-effective. 9 Machine learning,10–12 network analysis,13–15 and text mining16,17 are the major computational approaches that have been applied to problem solving. Furthermore, with the development of various machine learning and embedding techniques, multiple studies have evaluated their effectiveness in drug repurposing tasks. Donner et al. 18 have used deep neural networks to comprehend gene expression embedding profiles to predict pharmacological similarities for drug repurposing. Mei and Zhang 19 have proposed a multi-label learning framework to discover new uses for old drugs and new drugs for known target genes using the L2 regularized logistic regression. In addition, Xuan et al. 20 have presented a non-negative matrix factorization-based method to predict new indications for existing drugs using diverse information encompassing disease similarities, associations between drugs and diseases, and drug similarities. Moridi et al. 21 have extracted drug and disease features and represented them using deep learning to reveal their semantic relations in drug repurposing. These approaches have been applied to various types of data, including genomic,22–24 phenotypic,25–27 and chemical data.28,29
While previous studies have made great progress, two major limitations are worth considering. They either used limited types of data from a limited number of databases or failed to consider the interaction among multiple types of data. Data integration and representation are important to address these limitations. Through data integration, we can systematically gather data from multiple sources and store the obtained data in a central repository. Next, the data in the central repository can be presented in a format that can be directly used by computational approaches such as machine learning. Two major challenges need to be addressed for effective and practical integration and representation of data. The first challenge is the design of a unified data model incorporating multiple types of data from multiple databases. The unified data model should be general enough to embrace data from multiple databases and flexible enough to include database-specific information as well. The other challenge is the representation of data in a tabular format to be directly used as inputs of traditional computational approaches, while preserving the interaction among data as much as possible. Given that simple concatenation of different types of data and using them as features of machine learning approaches result in significant loss of connectivity information, effective data representation methods are of great need and importance.
In this study, we addressed the two aforementioned challenges using a comprehensive drug knowledge graph and data representation methods that consider the interaction among multiple types of data. Specifically, we have the following research questions:
How to systematically model and manage multi-source drug-related data?
How to represent the data in a format that can be used by graph-based approaches?
How to effectively combine data representation methods and graph-based approaches for drug repurposing?
To successfully address these research questions, we first construct a drug-centric knowledge graph that includes five types of entities: drug, disease, gene, pathway, and side effects, by integrating widely used drug knowledge bases. Then, we demonstrate path- and graph embedding–based data representation methods that can utilize the topology information of the drug knowledge graph. Finally, we demonstrate how the drug knowledge graph and the two data representation methods can be used along with the machine learning approach to predict drug repurposing candidates and explore a case study.
Methods
Drug knowledge graph
A knowledge graph is essentially a semantic network that reveals relationships between entities. One of the major concerns of a knowledge graph is the connectivity among the known facts stored in the knowledge base. By connecting pieces of knowledge gathered from knowledge bases, we obtain a more comprehensive and centralized repository. Therefore, constructing a knowledge graph allows the best use of previously discovered knowledge, revealing new knowledge based on the large-scale, interconnected known facts.
In the area of pharmaceutical research, tremendous human efforts have been devoted to curate drug-related knowledge. Hence, many drug knowledge bases have been developed and introduced to serve various academic research. Our previous work 30 has compiled a non-exhaustive list of widely used and publicly available drug knowledge bases. The study provides a detailed explanation of drug knowledge bases, including types of entities and relations, sources, and their applications in drug-related studies such as biomedical text mining, drug repositioning, adverse drug reaction, and pharmacogenomic analysis. From the available list, our drug knowledge graph was based on six drug knowledge bases, including PharmGKB, 31 TTD, 32 KEGG DRUG, 33 DrugBank, 34 SIDER, 35 and DID, 36 which have been selected based on the availability of raw data files. Figure 1 shows a few steps involved in the construction of the drug knowledge graph. As demonstrated, the extraction of structured information and the integration of the extracted information were the two major steps involved. In the following sections, we briefly summarize a few challenges encountered, as well as the approaches used to handle these challenges.
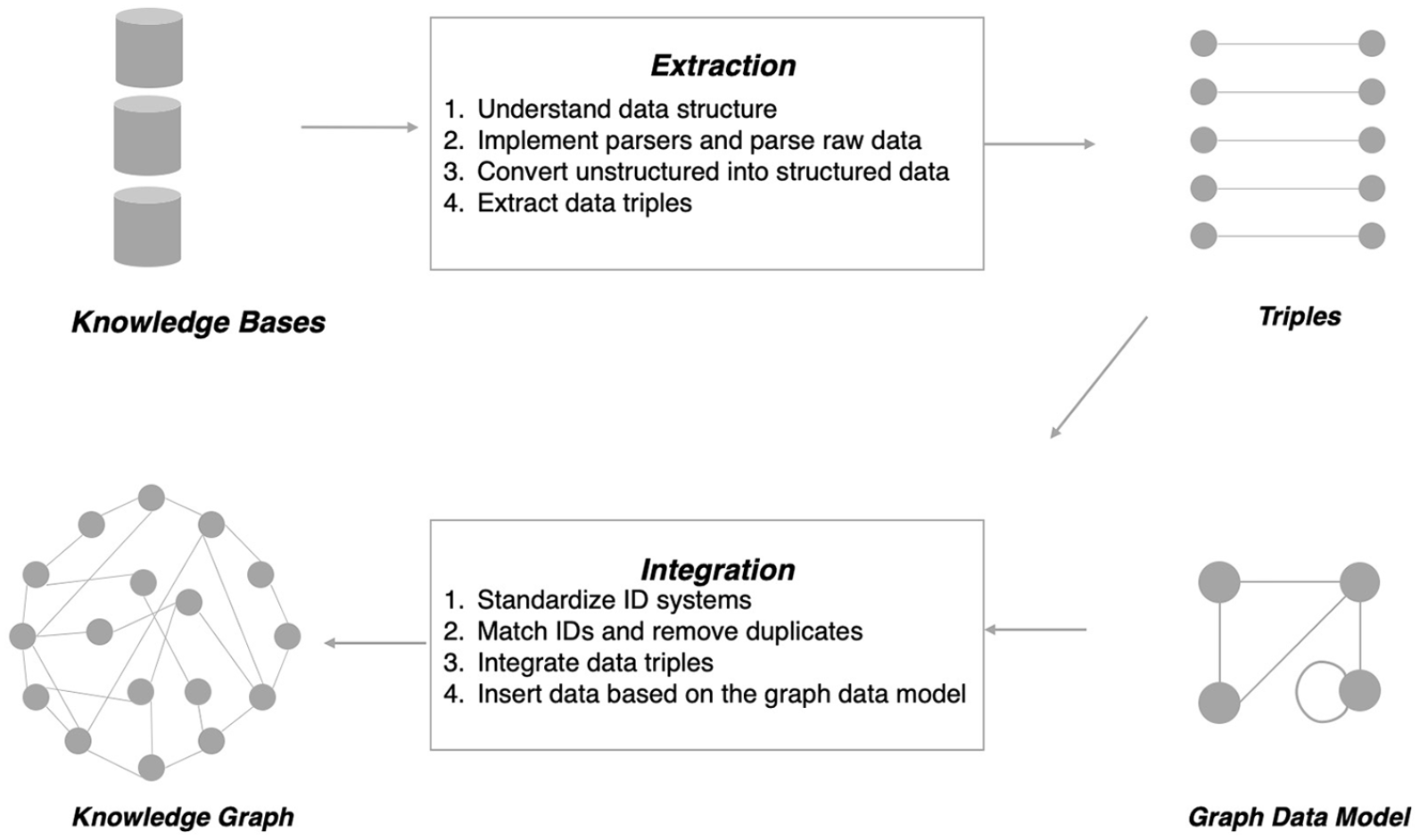
Major steps in the construction of a drug knowledge graph.
Extraction of structured information
Parsing of raw data files includes two important steps: (1) understanding structures and (2) implementing parsers. Drug knowledge bases distribute raw data files in various formats, including plain text, CSV, TSV, XML, and XLSX. In addition to direct download, REST API is another method to obtain raw data using HTTP requests. In most cases, structures of raw data are not explicitly defined, and we manually reviewed the data files to understand the structure. Plain text is the least structured format, and parsing it requires several trials and errors, testing different separators and regular expressions. Parsers vary by knowledge bases or even by files within the same knowledge base, and we implemented customized parsers based on the identified data structures.
Raw data include both structured and unstructured information. While structured information can be directly extracted and used without preprocessing, unstructured information should be manually reviewed and processed. For example, in some drug knowledge bases, diseases are provided with raw names with neither IDs nor cross-references to other ID systems. Since diseases are often described using several different names, direct matching only uses raw names and could induce uncertainties. For integration, these disease names need to be mapped to a universal ID system. As the universal ID system, we selected the Unified Medical Language System (UMLS). We combined both automated and manual methods to match raw disease names to IDs. Metathesaurus Data Files “MRCONSO.RRF” of UMLS consist of different names of diseases and their IDs, that is, Concept Unique Identifiers (CUIs). We found CUIs of diseases in the text by matching the string with the names in “MRCONSO.RRF” files. If the matching fails, we obtain CUIs by manual search on the website (https://uts.nlm.nih.gov/metathesaurus.html). Another widely used type of unstructured data is text. For example, in some databases, indications of drugs are described using long text information. With no certain natural language patterns used in the description, we manually extracted meaningful information.
Integration of the extracted information
Structured information extracted from the existing knowledge bases is fragmented. It is essential to integrate this information by connecting them based on a common medium and removing duplicates. We list below a few major issues during the integration.
The integration was based on cross-referencing. For example, if two records from two distinct drug knowledge bases have a common reference to another drug knowledge base, we integrated them into a single record in our drug knowledge graph. A simpler approach is considering cross-references only among the drug knowledge bases being integrated. However, this will result in a loose integration because many cross-references among the drug knowledge bases being integrated are missing and incomplete in many cases. In addition to the six drug knowledge bases, we also considered ID systems of other terminologies and knowledge bases. Here, a challenge was that the same ID system is referred to with different names in different knowledge bases and the formats of ID value also differed. For example, PubChem is used with varying names such as PubChem Compound, PubChem CID, PubChem ID, and CID. The values of PubChem ID are also described differently, that is, plain numbers or plain numbers followed by some characters. Therefore, we manually reviewed all the variants of ID systems used in the six drug knowledge bases. In total, we observed 25 distinct ID systems that had been used to describe the drugs.
One ID value of an ID system can match more than one ID value of another system. For example, a drug in DrugBank can match two UMLS CUIs. We manually reviewed many of these cases and found two main reasons for this one-to-many relationship: human errors and different levels of granularity. Since the development of knowledge bases requires significant human efforts, human errors are inevitable. When constructing a knowledge base manually, a researcher may assign wrong IDs to drugs and/or other entities. When an entity has inconsistent IDs in a knowledge graph, we believe it is a human error. For example, a drug in KEGG drug database has two different IDs, which also refer to two different drugs. As a result, some of the one-to-many relationships may include incorrect matching. Different levels of granularity is another reason for the one-to-many relationship. Drug knowledge bases describe entries (e.g. drugs) using different levels of granularity, and a higher-level entry in one drug knowledge base usually matches to many lower level entries in other drug knowledge bases. As it is impossible to manually review all the entries of one-to-many relationship to filter human errors, we maintained all of them. This decision was based on the result of our initial review of randomly selected entries, in which the dominant reason for the one-to-many relationship was noted as the different levels of granularity rather than human errors. Due to a large number of entries of the one-to-many relationship, if we failed to consider them and only integrate entries of a one-to-one relationship, the integration would be loose, which means the same entries may appear several times in the integrated drug knowledge graph.
Drug-centric graph data model
Data model is at the center of constructing a knowledge graph. A data model guides us regarding what and how data should be extracted and integrated. When designing a data model, we usually need to establish a compromise between domain knowledge and data availability. A perfect and detailed data model designed solely out of domain knowledge is usually unusable as data that fit the data model do not necessarily exist. When designing the data model, we accounted for a few factors. First, the data model should be general enough to embrace data from multiple sources. Second, the data model should be flexible to changes that might be needed when adding new data sources. Finally, and most importantly, interaction among multiple types of data should be properly represented. Based on these considerations, we propose a property graph model as shown in Figure 2. Our drug-centric graph data model comprised five types of entities and nine types of relationships among the five entities. The data model includes drugs, diseases, and other entities that interact with the two entities, such as genes, pathways, and side effects. The nine relationship types include TREATS (between drugs and diseases), INTERACTS (between two drugs), CAUSES (between drugs and side effects), BINDS, REGULATES, ASSOCIATES (between drugs and genes), ASSOCIATES (between two genes), ASSOCIATES (between genes and diseases), and PARTICIPATES (between genes and pathways). While most of the relationships are self-explanatory, we discuss some of these relationships that need further explanation. The BINDS relationship represents a drug binding to a protein encoded by a gene, REGULATES represents either upregulation or downregulation via the type property, and ASSOCIATES between a drug and a gene represents the mechanism of actions, such as activator or inhibitor. The graph data model serves as the guideline to link real biomedical entities and form a drug knowledge graph populated with instances. Therefore, in the drug knowledge graph, a drug instance can interact with various biomedical instances in terms of the abovementioned nine relationship types, and this rich information from the core of the drug knowledge graph.
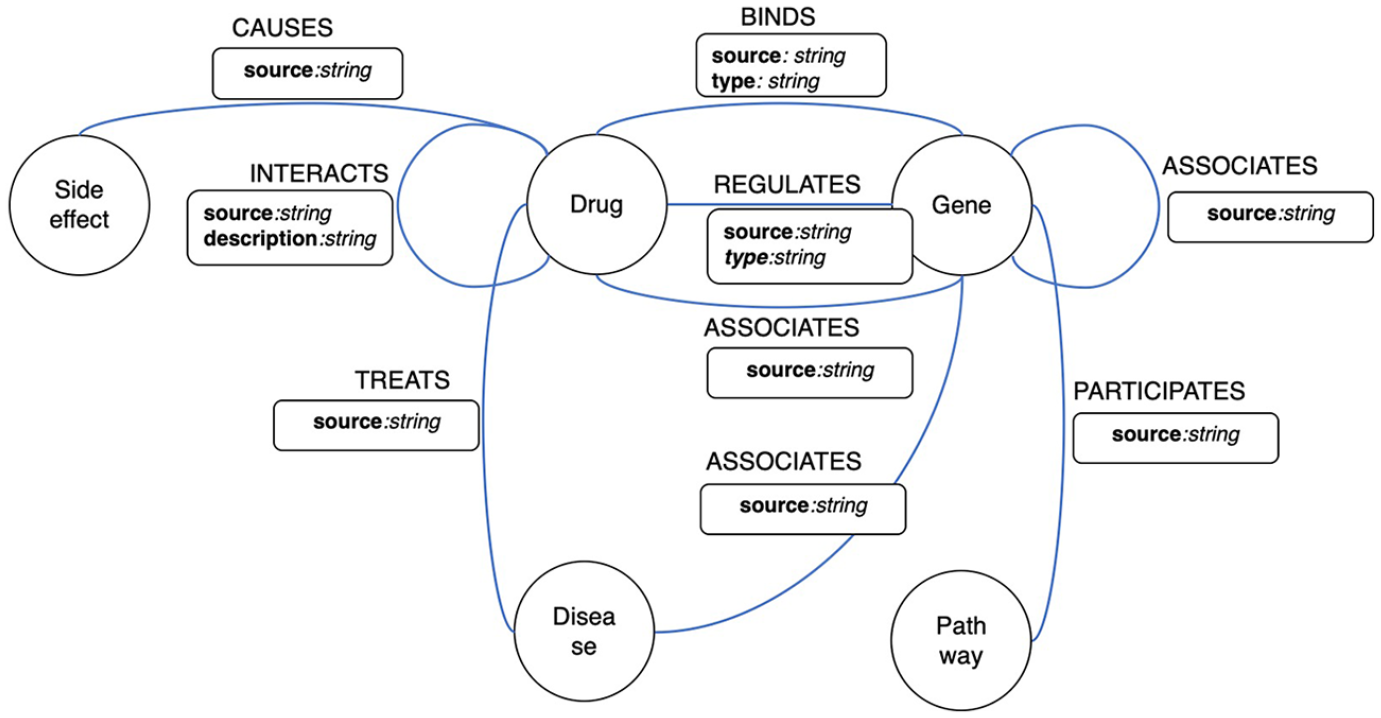
A drug-centric property graph model.
Data representation
Path-based representation
The two central entities in drug repurposing are drug and disease. How pairs of a drug and disease are connected in a drug knowledge graph provides significant information essential for uncovering drug repurposing candidates. Specifically, meta paths, which are sequences of relationships between two types of entities,37,38 are effective indicators. For example, based on Figure 2, many meta paths connect a drug with a disease. They include, but are not limited to, drug–gene–disease, drug–drug–disease, and drug–side effect–drug–disease. The meta path-based approach is able to capture local network structures around drug repurposing candidates. While it does not consider the whole network structure, only local structures (i.e. meta paths) that play important and intimate roles for drug repurposing candidates are considered.
Defining meta paths and measures of counting meta paths are the two important components of the approach, in which the path length affects the number of meta paths between two types of entities. In this study, we define all meta paths of length 2–4, decided based on both academic and practical considerations. Based on the model proposed in Figure 2, meta paths that are longer than four could lead to many uncertainties and thus it could be harder to interpret. For example, drug–drug–drug–drug–gene–disease, a meta path of length 5, includes three drug–drug interactions that could be interpreted in several possible ways. Practically, the number of meta paths increases exponentially as the path length increases. With this in mind, we propose all meta paths of length 2–4 (i.e. 99 meta paths) after considering relationship types and properties of relationships (e.g. upregulation and downregulation of REGULATES relationship).
We used the following four measures of counting meta paths, in which P denotes a meta path, p denotes an instance of the meta path, and h and t denote a head and a tail of a meta path, respectively
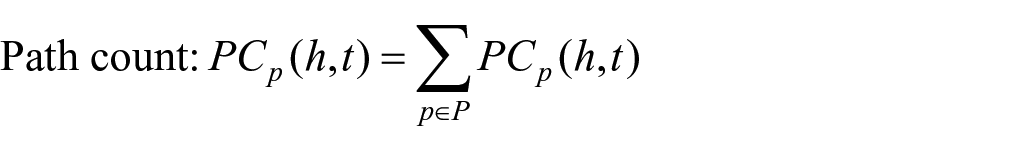
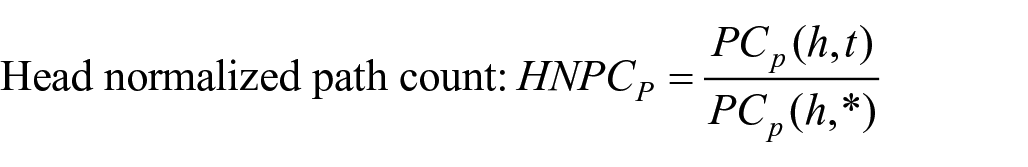
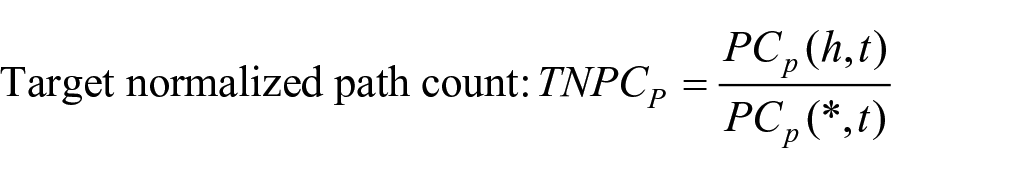
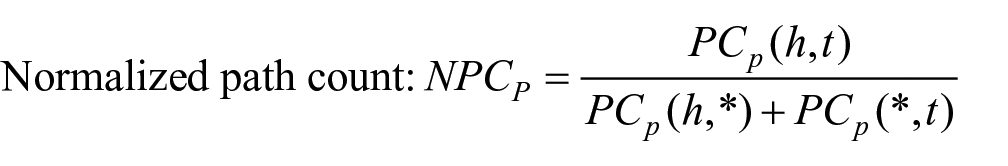
As shown above,
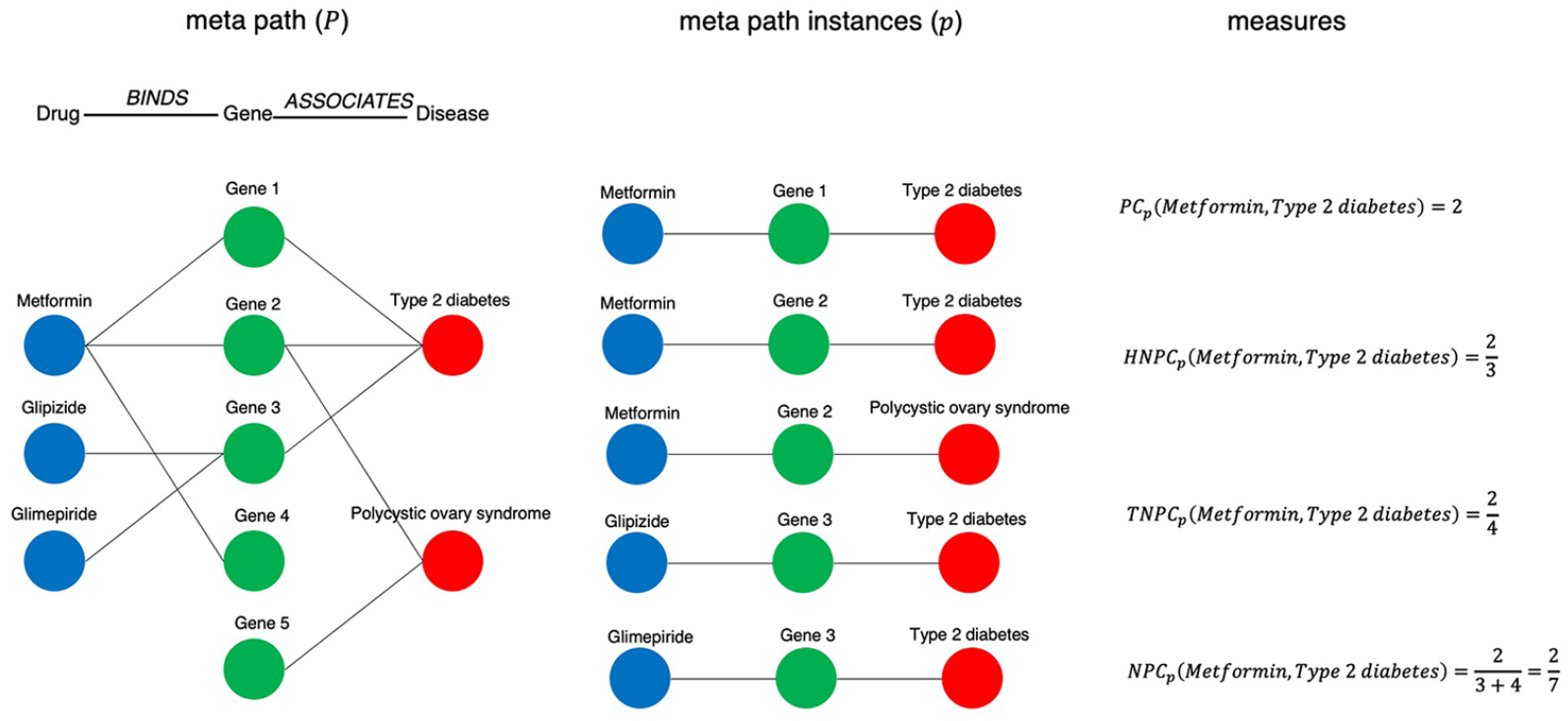
Examples of the path-based approach.
Embedding-based representation
Knowledge graph embedding 39 is a data representation method that represents entities and relations in vector spaces. Knowledge graph embedding possesses the advantage of projecting entities and relations in a knowledge graph in low-dimensional vector spaces by considering the topology of the knowledge graph. Therefore, the embedding-based representation is a global data representation method that considers all interactions among entities in a knowledge graph. Our previous study 40 surveys a list of graph embedding techniques in biomedical data science, including non-attributed network embedding and attributed network embedding. Their applications in biomedical data science, such as pharmaceutical data analysis, multi-omics data analysis, and clinical data analysis, have been discussed in detail. Recently, among the many approaches available, translation-based models have acquired extensive research attention. 41 Translation-based models, pioneered by TransE 42 and followed by variants, such as TransH, 39 TransR, 43 and others, translate an entities’ old vector representations into newer ones, in which the translations are performed by relations.
More formally, translation-based models share the following principle, in which

Finally, the goal of the translation-based methods is to minimize the following score function using the known facts, in which

The three methods mentioned above (i.e. TransE, TransH, and TransR) differ in the manner of translating
We illustrate differences between the three methods with an example in Figure 4. In the example, embeddings of metformin (drug), type 2 diabetes (disease), and TREATS are learned in different ways using different projection mechanisms.
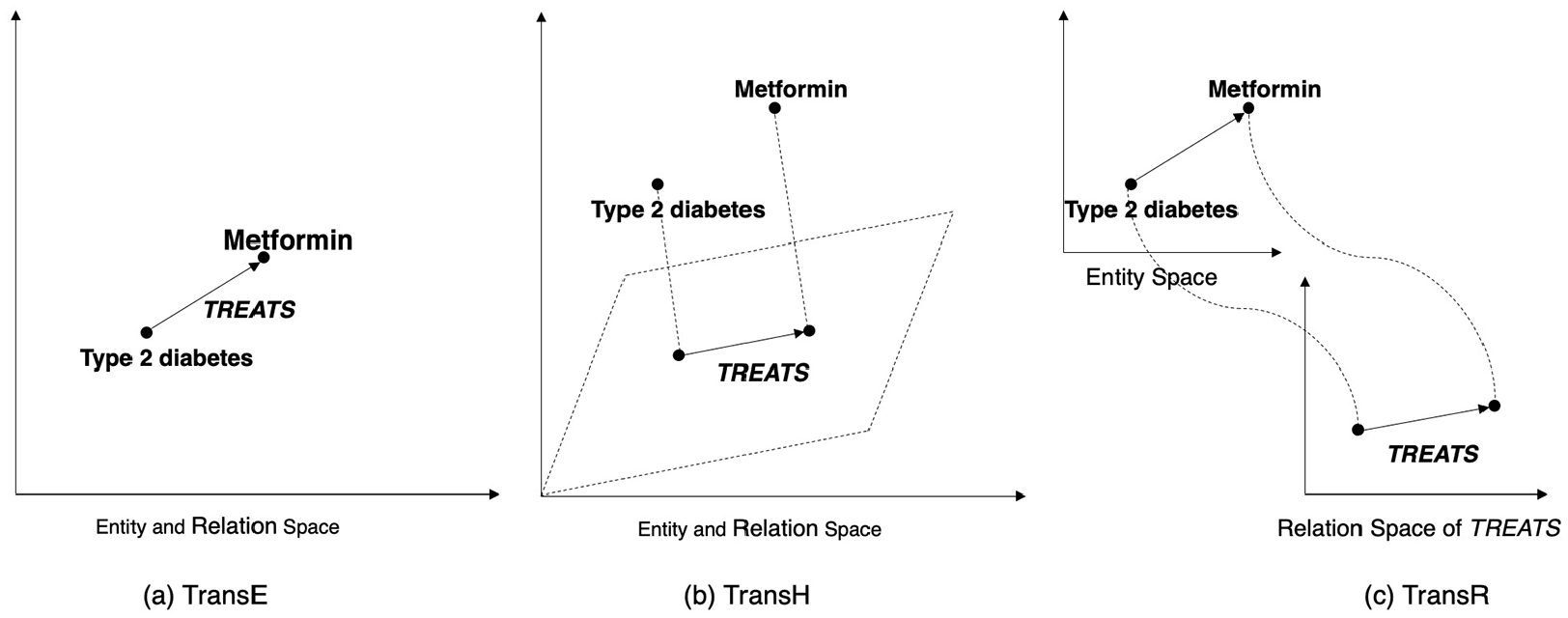
Illustration of the embedding-based approach.
After learning the embeddings of entities and relations, to find new indications for existing drugs, we represent drug–disease pairs of interest as
Results
Large-scale drug repurposing
In the experiments, we applied the above data representation methods to the drug knowledge graph and generated feature matrices to train machine learning models. As we only had positive samples, that is, known treatments, when training machine learning models, we did not possess inputs for negative samples. Instead, we had unlabeled samples whose labels (either positive or negative) were unknown. Therefore, a class of machine learning approaches called positive and unlabeled (PU) learning, 45 using only PU data to train machine learning models, was used. Unlabeled samples usually provide additional information to machine learning models for the purpose of learning. For the experiments, we used the approach proposed by Elkan and Noto, 46 which has been widely used in the previous studies. 47 The method uses a traditional machine learning algorithm to first predict labels for the unlabeled data and then assumed the prediction results as the correct labels to train another machine learning model. We implemented a PU learning method with three machine learning algorithms (i.e. Decision Tree, Random Forest, and support vector machine (SVM)) available in the scikit-learn package. Decision Tree and Random Forest employed Gini impurity to measure the quality of a split. Random Forest built 100 trees in the forest with bootstrapping samples. SVM used radial basis function (RBF) kernel and enabled probability estimates.
Since the size of all unlabeled samples (drug–disease pairs), obtained by generating all possible pairs of drugs and diseases that do not have known effects, is huge and it is computationally expensive to obtain all of them, we focused on diabetes mellitus and included unlabeled samples that were related to the disease. The goal of the experiments was to evaluate the effectiveness of the drug knowledge graph and the various data representation methods in terms of predicting known diabetes mellitus treatments based on the known effects of other drug–disease pairs. Based on the MeSH tree numbers of diabetes mellitus (C18.452.394.750 and C19.246) and MeSH IDs of diseases in our drug knowledge graph, we identified eight diseases (diabetes mellitus, type 1 diabetes mellitus, type 2 diabetes mellitus, diabetic nephropathies, diabetic retinopathy, diabetic neuropathies, Wolfram syndrome, and Donohue syndrome) that fall into the category of diabetes mellitus in the drug knowledge graph.
As shown in Figure 5, we trained two classes of machine learning models, one for path-based and the other one for embedding-based.
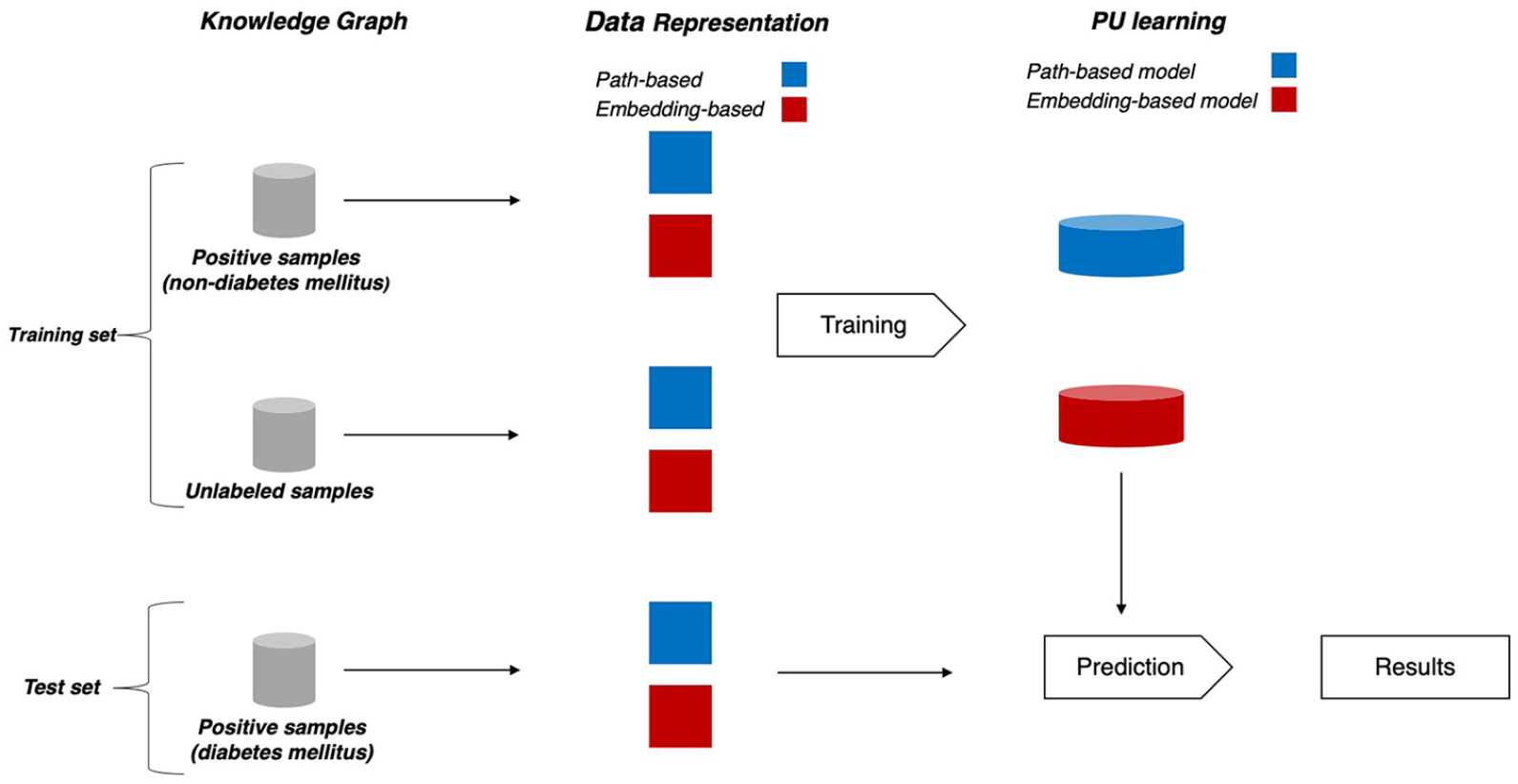
Machine learning pipeline.
In the experiments performed, our test set comprised all known treatments of the eight diseases. All known treatments of the other diseases constituted our training set together with 143,830 unlabeled samples, obtained by associating the eight diseases with drugs that treat at least one disease. Table 1 shows the size of the training and test sets.
The size of the training and test set.

As our test set included only positive samples, in Table 2, we report recall (or sensitivity) that measures the proportion of known treatments that were correctly predicted.
Recall scores for the data representation methods.
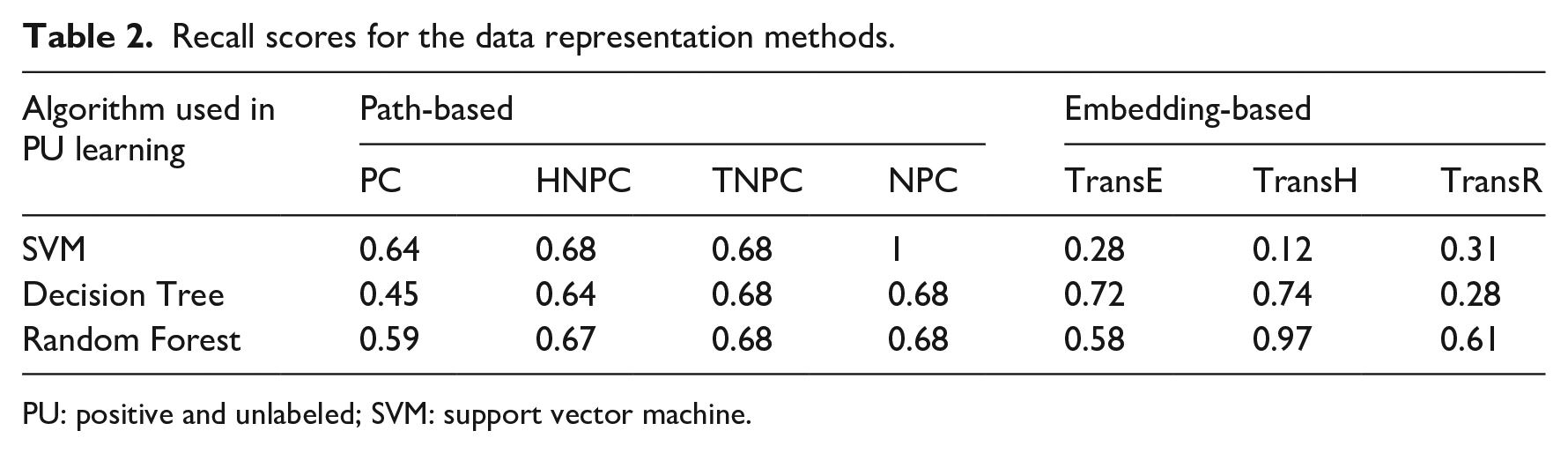
PU: positive and unlabeled; SVM: support vector machine.
The performance varied by different combinations of the data representation methods and machine learning algorithms. As shown in Table 2, with certain specific combinations, we observed perfect (SVM + NPC) or nearly perfect (random forest + TransH) prediction results.
A case study
Using the path-based approach, we can explore a specific drug–disease pair in terms of their connectivity to understand the possible treatment relationship between the two. While the machine learning approach is more suitable for large-scale prediction, path-based exploration can deliver additional interpretability and insights about a specific case. Metformin, a widely used antidiabetic medication, has been widely studied as an effective antineoplastic medication. Studies have investigated its effects on various types of cancers, including gastrointestinal cancers, breast cancer, prostate cancer, ovarian cancer, and lung cancer. 48 Recently, a study reported that metformin may improve chemotherapy outcomes in lung patients with diabetes. 49
Evaluating the meta paths that connect metformin and lung cancer is an effective method to explore how metformin and lung cancer interact with each other and what other entities (e.g. genes) play roles in associating the two. The 99 meta paths (length 2–4) mentioned earlier can be grouped into eight types, if we do not consider the types and properties of relationships and broadly treat them as interaction or association. Table 3 shows the eight types and the counts of instances for each type that connects metformin with lung cancer.
Meta paths connecting metformin with lung cancer.
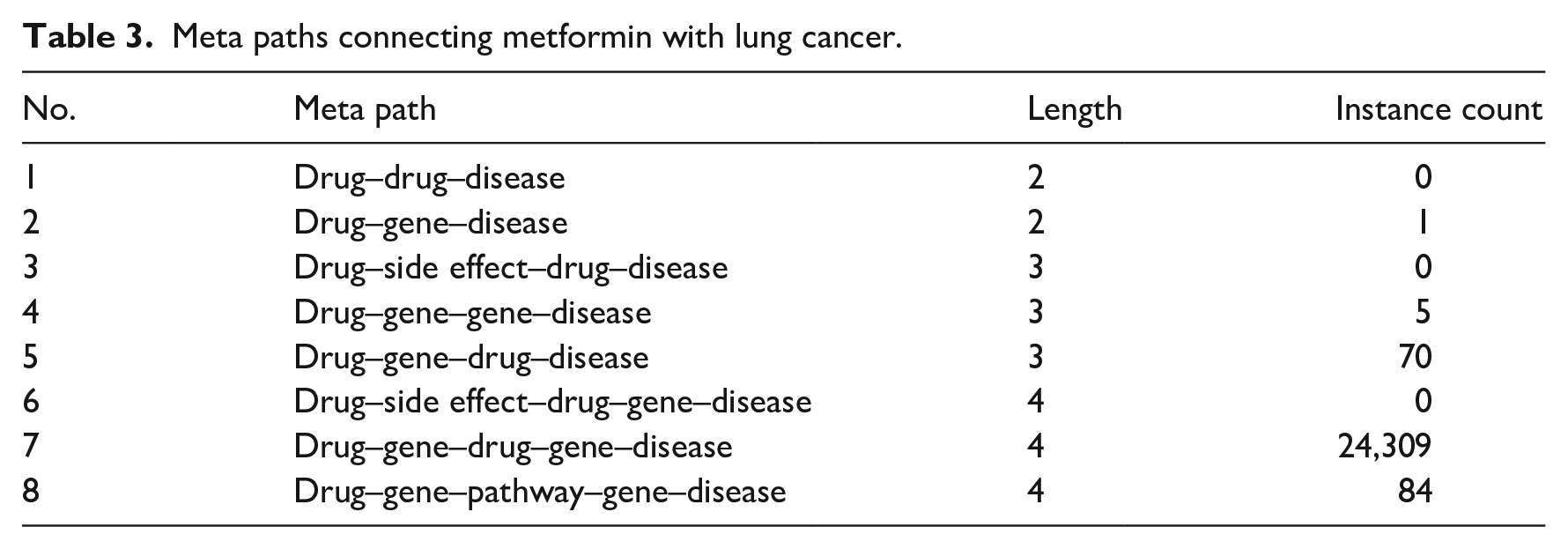
In Table 3, the first drug in each meta path denotes metformin and the last disease denotes lung cancer. Some meta paths demonstrate zero instances, implying that there were no actual paths of these kinds connecting metformin with lung cancer. The fourth meta path, drug–gene–gene–disease, can be interpreted as two genes that interact with metformin and lung cancer separately and are associated with each other. Therefore, the association between the two genes might be a useful piece of information that helps understand the possible treatment relationship between metformin and lung cancer. Figure 6 shows the five instances of drug–gene–gene–disease that connects metformin with lung cancer. Genes that interact with metformin and/or lung cancer are plotted along with their association.
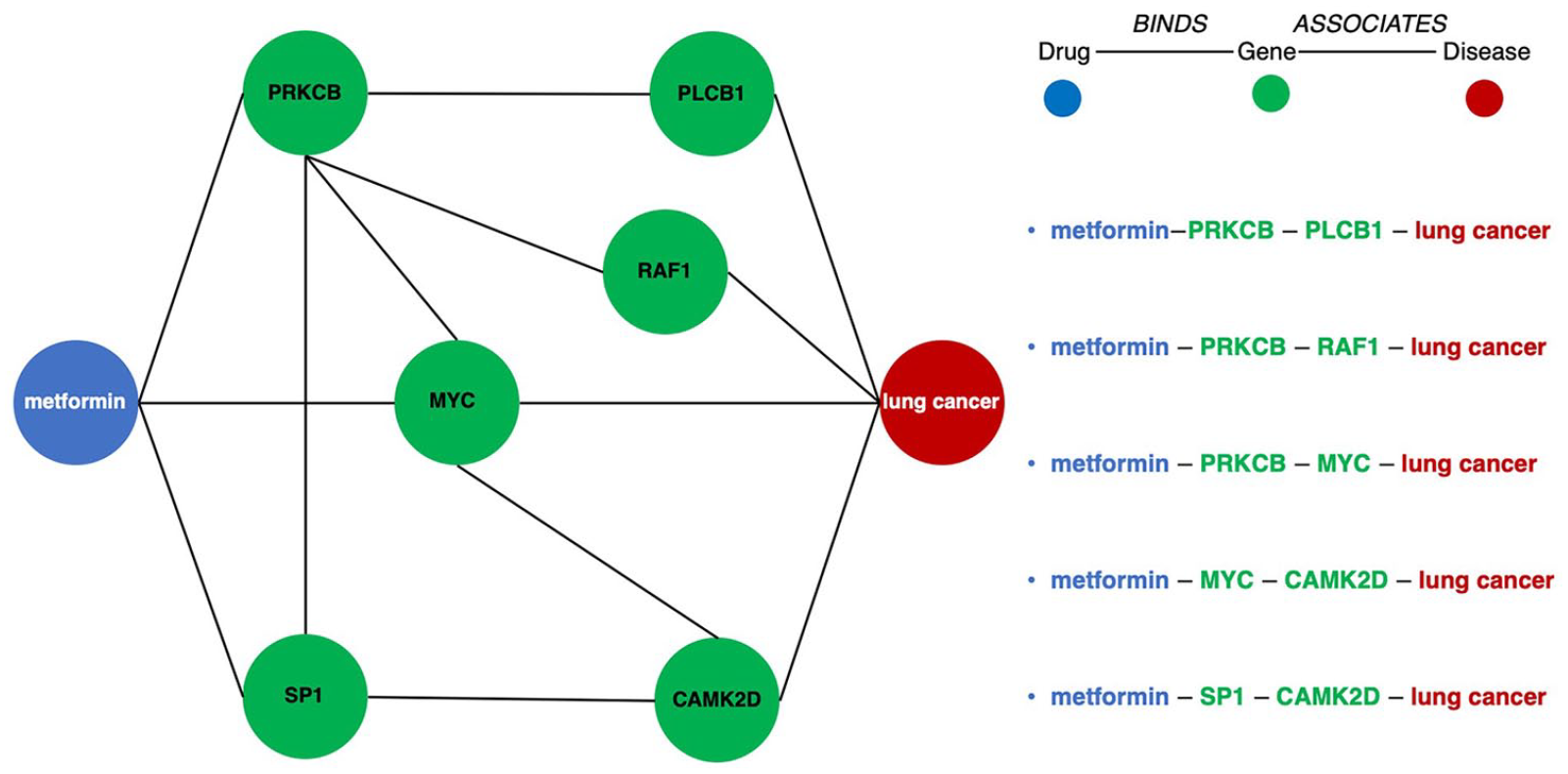
The five instances of drug–gene–gene–disease that connect metformin with lung cancer.
The five meta path instances shown in Figure 6 are a part of a regional network that consists of more than 20,000 meta path instances (sum of the last column in Table 3), connecting metformin with lung cancer. Each meta path instance conveys a useful piece of information, and therefore, path-based exploration can be used as either a follow-up to machine learning-based prediction to discover evidence or as a mean to broadly explore mechanisms of drug action.
Discussion
Based on our results, we observed that the drug knowledge graph introduced in the study provides sufficient background information for drug repurposing. The drug-centric property graph model was able to capture the general and essential information, revealing multiple aspects of the drug and its interaction with other biomedical entities. Due to the proposed drug-centric property graph model, we were able to systematically extract and integrate data of six drug knowledge bases and construct the drug knowledge graph. The path-based data representation method was able to provide local, yet very intensive information about interactions between drugs and diseases, while the embedding-based data representation method provided global and comprehensive information. Both methods applied to the drug knowledge graph produced high predictive results on diabetes mellitus treatments with certain machine learning models, while only using treatment information of other diseases.
This study has a few limitations. First, despite our dedicated effort to construct the drug knowledge graph, we were not allowed to make it publicly available due to restrictions on data republishing asserted by some of the drug knowledge bases. However, we expect, with the drug-centric property graph model and the approaches we described in this study, researchers would be able to construct a comparable graph for their own studies. Second, since obtaining data representation matrices for all unlabeled examples is computationally expensive and time-consuming, our evaluation focused only on a specific disease with a reasonable number of unlabeled examples. Nevertheless, the proposed approaches can be applied to other diseases. Third, in terms of the evaluation metrics, since we only had positive samples in the test set, only recall was reported. If negative examples were available, a more multifaceted evaluation would have been possible.
Conclusion
Prior knowledge regarding various aspects of drugs is the key to successful drug repurposing. Constructing drug knowledge graphs is an essential step to obtain the best use of prior knowledge by weaving the continuously growing, fragmented, and dispersed drug-related data. By applying effective data representation methods to the drug knowledge graph and thus transforming knowledge available in the drug knowledge graph into informative inputs for machine learning models, we can effectively predict drug repurposing candidates. Large-scale prediction and interpretability are well-known limitations of experimental approaches and previous computational approaches, respectively. The knowledge-driven approach supports not only large-scale prediction through data representation and machine learning methods but also allows investigation of a case study through path-based exploration. These two features can effectively handle the abovementioned limitations with the support of a comprehensive drug knowledge graph.
Footnotes
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by the Ministry of Education of the Republic of Korea and the National Research Foundation of Korea (Grant No. NRF-2019S1A5A8033338).