
Abstract
The European Union Medical Device Directive 2007/47/EC1 defines software with a medical purpose as a medical device. The implementation of health information technology suffers from patient safety problems that require effective post-market surveillance. The purpose of this study was to review, classify and discuss the incident data submitted to a nationwide database of the Finnish National Competent Authority with other forms of data. We analysed incident reports submitted to the authority database by users of electronic health records from 2010 to 2015. We identified 138 valid reports. Adverse events associated with electronic health record vulnerabilities, clustered around certain error types, cause serious harm and occur in all types of healthcare settings. The low rate of reported incidents raises questions about not only the challenges associated with medical software oversight but also the obstacles for reporting.
Introduction
Historically, regulation governing Health Information Technology (HIT) has been less strict than for medical devices. 1 Today, however, systems in both the European Union (EU) and the United States use models for reporting failures and adverse events related to medical device–related software, but the systems differ.1–5 EU legislation has for some years already regarded software as a medical device. Directive 2007/47/EC 6 amended the definition of the term ‘medical device’ used in Directive 93/42/EEC, 7 subjecting stand-alone software with a medical purpose to oversight under the medical device directive.
In 2016, the European Commission published guidelines known as MEDDEV 2.1/6 8 on the qualification and classification of stand-alone software when used in a healthcare setting. The EU directive requires the achievement of specific end results, and implementation of the directive must adhere to national regulations. 6 The primary purpose of the EU Medical Device Vigilance System is to improve patient and user health and safety protections reducing the likelihood that incidents would recur. 7 The US system has no regulatory requirements to evaluate electronic health record (EHR) system safety 9 and does not systematically or consistently track adverse outcomes associated with EHRs. Regulatory data are stored in several large Food and Drug Administration (FDA)-supported databases. 4
An Institute of Medicine 10 report found that an increase in the implementation of HIT could potentially lead to patient safety incidents by introducing new vulnerabilities. Evidence of unintended consequences has grown over the past decade, but is limited, as demonstrated in a systematic review of eHealth technologies and their impact on healthcare safety. 11 A recent Finnish study in a setting with a 100 per cent implementation rate for EHRs found significant problems with their use.12–13
Only a few studies have focused specifically on HIT supervisory data outside the EU. 14 The EU totally lacks study results in oversight from the user’s perspective. The United States, in contrast, has seen the publication of several valuable expert opinion papers about software oversight.2,4,5
The regulatory oversight system should gather data to help HIT developers and clinicians more fully understand and mitigate risks associated with HIT implementation and use. The existing infrastructure for patient safety reporting and analysis is currently unable to quantify the rate of HIT-related patient safety events with any precision.2,15 Among its critical limitations is a lack of common, easily usable preferably EU-wide IT-specific taxonomy for errors.16–17 Applying a taxonomy would facilitate one to capture and distinguish different types of failures and their causal factors 18 as part of preventive actions of HIT safety. Another crucial limitation of existing HIT-related systems for reporting safety events that deserves more attention is that most clinicians do not understand what they should be reporting.5,15
The overall purpose of this study was to review, classify and discuss the incident data submitted to a nationwide database of a national competent authority (Valvira) in Finland. This study seeks to contribute information on EHR-related problems by using commonly identified error types based on user- and system-related sociotechnical factors 13 in professional user reports.
Methods
Context
Finnish EHR systems cover 100 per cent of specialised and primary healthcare organisations. IT systems are usually developed or customised locally, but systems developed by multinational companies are also in use. Primary and specialised care organisations have integrated physician order entry with clinical decision support and major ancillary systems, a picture archiving and communication system, as well as a clinical data repository for reviewing results. The closed loop medication system is seldom part of the EHR system. A new version of EHR programmes was implemented in order to incorporate the systems into the Finnish National Health Care Archive KanTa in 2014–2015.12,13,19,20 Finnish healthcare organisations base their patient safety work on obligatory systems for reporting patient safety incidents (even though the reporting is voluntary for an individual professional), and complementary internal rules and regulations, in addition to legislation, guide medical device safety.12,21
To conduct our study, we used the database of the Finnish Regulatory Authority (Valvira), a national agency operating under the Ministry of Social Affairs and Health charged with supervising social and health care. Enforcing the EU legislation on medical devices is the responsibility of the authorities in each EU country, and incidents must be reported to the national Competent Authority in the country where the incident occurred. Valvira has issued regulations on reporting serious adverse incidents with medical devices for users and manufacturers since 15 September 2010. Serious adverse incidents are to be reported within 10 days of the user or manufacturer’s first awareness of the incident. This includes device deficiencies that led or might have led to a death, led to a serious deterioration in health or led to foetal distress, foetal death or a congenital abnormality or birth defect. Other incidents are to be reported within 30 days; failure to report is a criminal offence. Valvira provides a common template for reporting purposes. Documentation of medical devices from both EHR users and HIT vendors are stored in a national database reflecting risks in both hospital and community environments. 21
The National Supervisory Authority of Welfare and Health (Valvira) granted permission (Valvira no. 2849/06.01.03.01/015) for this register study in June 2015. We collected the data confidentially. The data analysis used only general definitions, thereby guaranteeing the anonymity of the content of the reports.
Methodology
A retrospective study of incident reports by EHRs users was conducted for cases submitted to the competent authority database during the period September 2010 through September 2015. This article reports on a descriptive, quantitative study using the quantitative content analysis approach 22 to HIT-related problems by applying commonly identified error types based on user- and system-related sociotechnical factors. The coding in this study is based on Sittig and Singh’s study findings and described in detail in previous research papers.13,23,24 The coding framework follows the structure of the FIN-TIERA (Finnish technology induced errors’ risk assessment) tool which consists of eight main categories: 25 EHR Downtime; System-to-System Interface Errors; Open, Incomplete or Missing Orders; Incorrect Identification; Time Measurement Errors; Incorrect Item Selected; Failure to Heed a Computer-Generated Alert; and Failure to Find or Use the Most Recent Patient Data.
The researcher received a list of report IDs which had been classified with an electronic patient record (EHR) tag related by the authority and thus belonging to the research focus. The list contained a total of 365 EHR-related reports during this period, including not only incident reports by EHR users but also National Competent Authority Reports (NCAR), Field Safety Corrective Action (FSCA) documents and other manufacturer reports. The study focused on EHR user reports only, with the exclusion of all other types of documentation. Additionally, in order to verify the list of report IDs containing all relevant user cases, we selected all Medical Device reports in paper format in the authority archive during the study period for the preliminary analysis: we checked every report to confirm whether it was EHR related and compared it to the list of report IDs. We noticed that three user incident reports went missing from the original report list as a result of the authority’s efforts to transition towards e-archiving; these were added to the list.
We continued the analysis by selecting all relevant EHR user reports for a subsequent in-depth content analysis. We removed from the research database any irrelevant cases that failed to meet the EU reporting criteria. These cases included reports concerning software not classified as a medical device, but rather for medication logistics, and one report classified as user feedback. We consulted the authority in borderline cases where the reporting criteria were unclear. We found no duplicate reports among the user incident reports.
It is especially noteworthy that the authority database also includes EHR user incident reports of health information system downtime. Downtime does not fall under the reporting criteria of the directive,6–8 but is instead a borderline issue for reporting, even though downtime incidents may seriously affect patient safety in institutions with a 100 per cent EHR implementation rate. We decided to include these cases in the analysis due to their importance for patient safety.
At the beginning of the coding process, the researcher pilot-tested the coding scheme with 30 reports. We identified relevant factors in each report based on a complete review of the national authority report file, which included the original reports and summaries as well as supplementary material. The authors, who were all familiar with the coding themes and content, discussed the coding rules several times. Presumably, the data may have contained reports that did not fit into the coding framework. Consequently, we decided to add an ‘Other’ class to the coding framework and to discuss the development needs of the classification later.
The first author (S.P.) then completed the coding independently. In the end, the co-authors discussed the coding principles once more and deemed the researcher’s decisions acceptable and according to the rules of the coding framework. The second author (K.S.), a professor of health informatics, coded the data independently; the results were discussed in detail. All the reports were coded according to precisely the same codes as the first coding by S.P. Kappa coefficients for computing inter-rater reliability were therefore deemed unnecessary because they were in perfect agreement.26–27 It is noteworthy that the authors have developed and tested the coding framework13,25 during the last 2 years which facilitates the interpretation and correct use of the framework.
After discussing the coding results, the data underwent additional analysis. Adding a dimension to the threefold taxonomy of the Agency for Healthcare Research and Quality (AHRQ) Common format 1.2 28 facilitated the identification of characteristics of certain content related to error type, thereby separating the adverse events resulting from medical devices into three classes: (1) device defect or failure, including HIT; (2) error in use; and (3) combination or interaction of device defect or failure and error in use. We used the authority’s remarks about the root cause of the incident in this analysis.
After the analysis of cases submitted to the competent authority database, we compared the results to the characteristics found in two previous studies carried out in Finland.
Results
Description of the error types
We analysed a total of 138 user incident reports, which included seven of the nine error types found in our previous study. The most common error types in the reports were (n = 37, respective) ‘Downtime’ (26.8%) and ‘System-to-System Interface Errors’ (26.8%). The error type ‘Open, Incomplete or Missing Orders’ accounted for 23.9 per cent (n = 33) of incidents and ‘Incorrect Patient Identification’ for 10.1 per cent (n = 14). The error types ‘Translational Challenges with EHR Time Measurements’ (2.2% per n = 3) and ‘Incorrect Item Selected from a List of Items’ (0.7% per n = 1) were among the least reported incidents.
Altogether 13 reports were classified under the error type ‘Other’, accounting for 9.4 per cent of incidents. The cases in this class did not fit into any other categories of commonly recognised EHR error types. These reports included five reports related to data security and the professional rights of licenced clinicians. 25 Professionals who should not have had access were able to view the data. In three cases, the software module specifically related to the administration of medication was locked so that clinicians were unable to view or handle the data in this module. Four reports detailed specific problems with the dictation module in the medical software that caused the application to add patient information which the physician did not dictate. One of the cases involved several error types, but failed to provide enough detailed information to reliably select the major error type.
The sample did not include the error types ‘Failure to Heed a Computer-Generated Warning or Alert’ and ‘Failure to Find or Use the Most Recent Patient Data’. Errors in 31 reports (22.5%) were related to the software used in the ePrescription modules implemented as part of the national ePrescribing system. Table 1 provides a summary of the descriptive statistics by error type.
Descriptive statistics (% per n) of error types in EHR user reports.
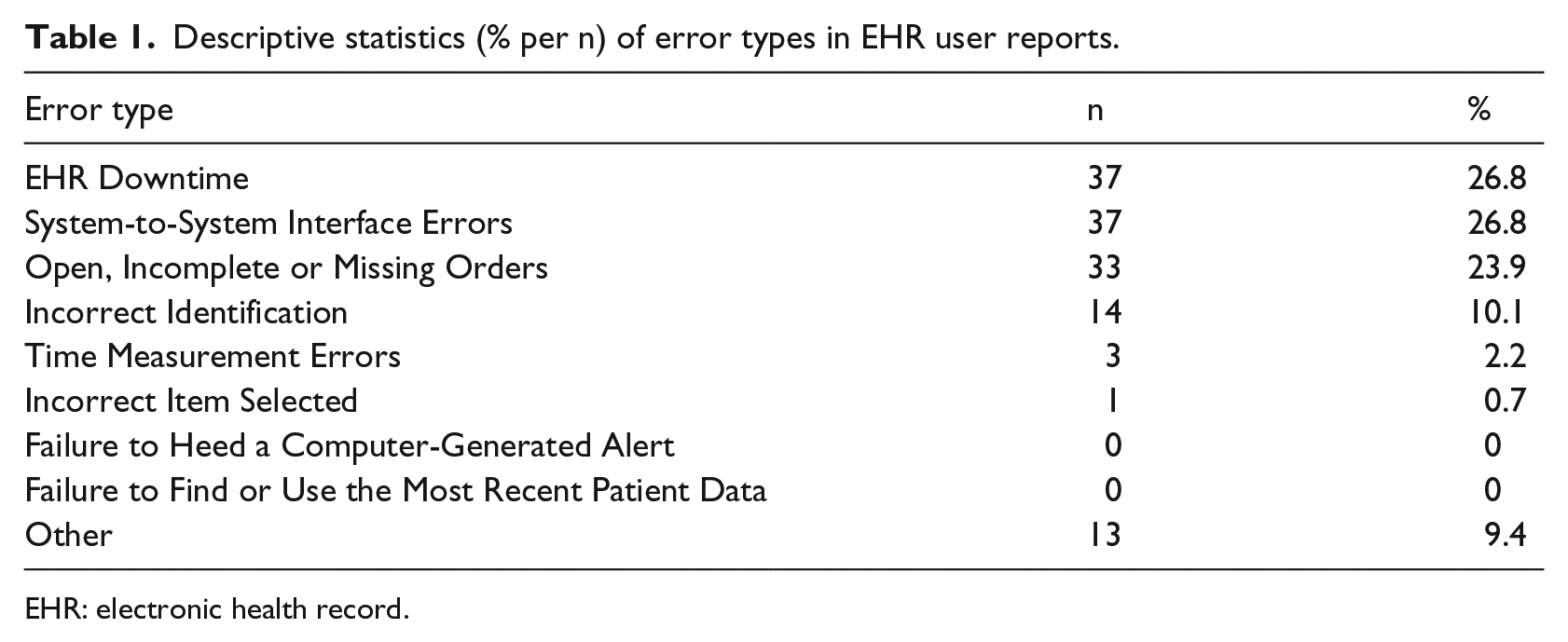
EHR: electronic health record.
Severity of the incident
Altogether 23.2 per cent (n = 32) of the reports were labelled as serious incidents. Of these, the competent authority rated 31, and the researcher who used supplementary information from the manufacturer’s report, which could be linked to the user reports, rated 1. Serious cases were typically related to prolonged EHR downtime and medication-related software errors. One serious case was related to an incident where a physician had discontinued a patient’s medication, but the software continued the medication with a higher dose than before. The only report in the error category ‘Incorrect Item Selected from a List of Items’ was a serious one: the software facilitated the selection of the wrong medication from a list, which led to serious consequences for the patient. The number of error types related to cases which were labelled as serious incidents is shown in Figure 1.
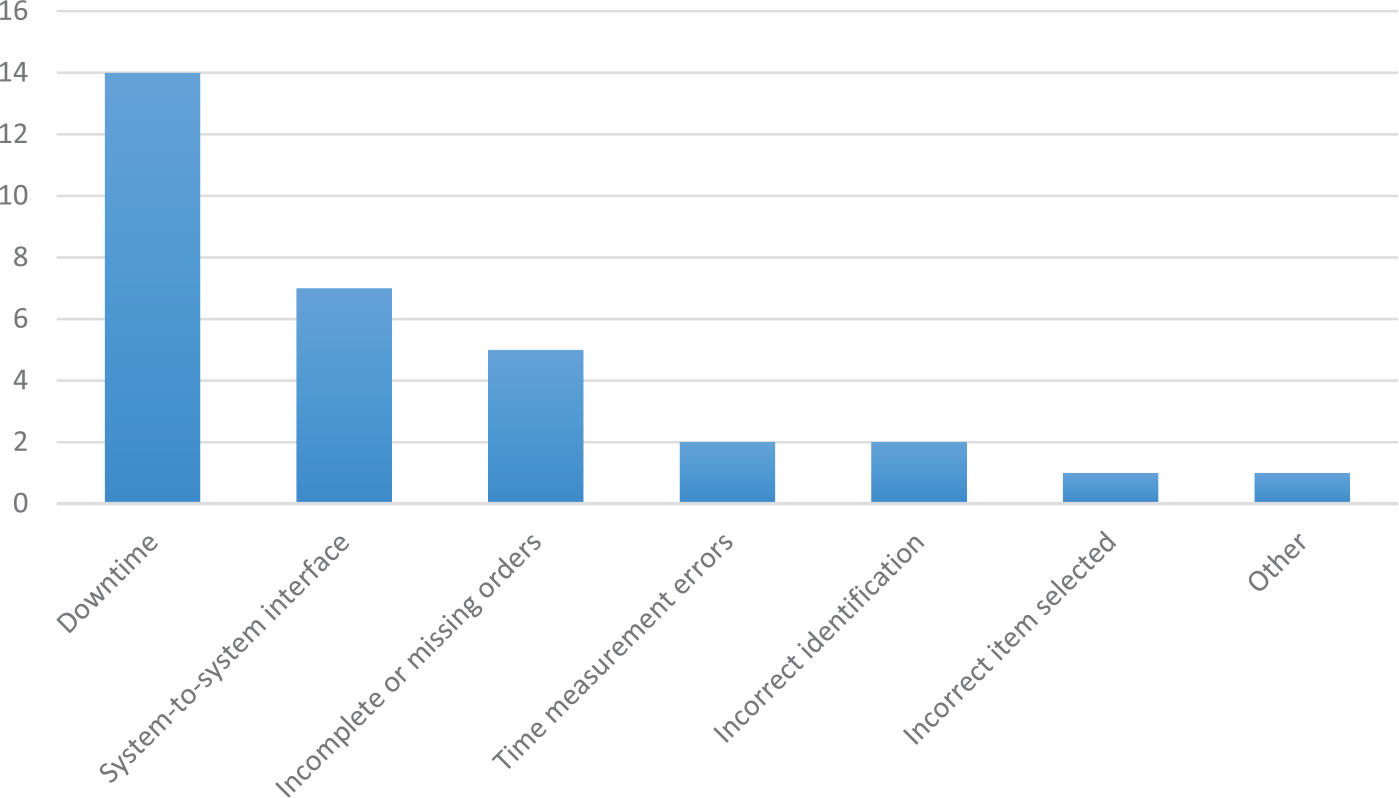
Descriptive statistics (n) of error types in serious EHR user reports.
Organization type
Primary care organisations, such as primary healthcare centres and community-elderly care homes, accounted for 38.5 per cent of cases (n = 53 cases). Specialised care organisations (e.g. university hospital districts and central hospitals) accounted for 58 per cent of the reports (n = 80). A private care organisation, such as a private doctor company, created two user reports. In three cases, the information available was insufficient to determine the type of organisation.
Remarks about the possibility of an error in use
After analysing the error types, we used the threefold taxonomy of the AHRQ Common format 1.2 to view the entire dataset. The results shown here are only directional due to the type of data (i.e. register data that did not allow a full investigation of the root cause in all cases).
These data include a total of 111 reports out of 138 reports in the class ‘device defect or failure, including HIT’ diagnosed by the competent authority. The authority labelled the majority of downtime-related reports, as well as System-to-System Interface Errors, as device defects or failures. We classified an additional 14 downtime and interface reports that lacked root cause analysis by the authority as a ‘device defect or failure’. In summary, a total of 126 reports (91.3%) involved device defects or failures.
According to the authority’s assessment, two reports contained the class ‘error in use’ with no device defect as the root cause. Both of these are prescription-related errors. Analysis revealed that two reports contained the class ‘combination or interaction of a device defect or failure and error in use’ in addition to patient identification errors. One of these two reports described several usability issues.
Discussion
Current EU directives governing medical devices and the requirement of member states to appoint a competent authority responsible for implementing these rules on the national level indicate a gradual move towards stricter regulation of medical software. At the moment, the EU is expected to propose new Regulations on Medical Devices 29 that will most probably lead to tighter surveillance. This is a reasonable and most welcome tendency since more serious EHR-related safety events are likely to occur as more organisations implement comprehensive EHRs. 5 This study also provided examples of risks stemming from the development or use of EHRs in environments with a 100 per cent implementation rate.
The results of a recent questionnaire study of nearly 3000 clinicians in Finnish hospitals supports the analysis in this study. The highest proportion – nearly half of the respondents – reported a high-risk level related to extended EHR unavailability, and the lowest overall risk level was associated with selecting an incorrect item from a list of items. 13 Even if system downtimes do not result predominantly from software problems that fall under EU directives, they do have a significant impact on HIT safety and require oversight measures. This study, like previous studies on EHR safety, stresses the importance of contingency plans in case of EHR downtime. We recommend that EHR downtime fall under the EU directive in a detailed manner so as to enhance wide-scale preparedness for these serious situations. When comparing the severity of incidents in this study, a previous study using the same kind of regulatory database found 11 per cent of 436 events associated with patient harm and 9 per cent of deaths were linked to HIT problems. 14 In this study, the number of severe cases exceeded 20 per cent, even though not all of these led to death, which further underscores the importance of fostering EHR safety procedures.
The obvious problem of underreporting in this study raises questions about the adequacy of the present supervisory system, which demands more attention. Two recent Finnish studies12–13 in a specialised hospital environment show that, if the supervisory system were working adequately, the number of reports in this study would be significantly higher on the national level. An important question is whether the Medical Device Vigilance fulfils its principal purpose to improve patient and user health and safety protections by reducing the likelihood that incidents would recur elsewhere. 7 This phenomenon is similar in the US system, which maintains only a small number of incidents in different databases. 5 The problem of underreporting has been recognised in an Australian study. Only 1.3 per cent of clinically important errors, with the potential to cause patient harm, were reported to the hospital incident systems. Almost 80 per cent appear to have failed to be detected by the staff. 30 Lack of feedback from incident reporting has been recognised to decrease the willingness of staff to report safety problems. Research predominantly done during the 2000s has identified several reasons that inhibit reporting, such as fear of blame, resource constraints and a lack of clear definitions of reporting criteria.31–33 First, greater attention should be placed on an increase in incident reports which should be encouraged and viewed as an indicator of an open and safe reporting culture. 30 Second, to learn from failures, people need to be able to talk about them without fear of punishment. Managers have an essential role: by reframing workers’ perceptions of failures from sources of frustration to sources of learning, managers can engage employees in system improvement efforts that would otherwise not occur. 34
The Medical software directive is a relative new for EHR users and healthcare management. Although the actual requirements of the directive remain unclear, the new MEDDEV guidelines 8 will likely improve the situation. The classification of software has been clarified even if the underlying principles of the directive remain unchanged. Stand-alone software must have a medical purpose to qualify as a medical device. However, not all stand-alone software used in healthcare settings should qualify as a medical device. Electronic patient records themselves, for instance, are not computer programs, but the modules used in an electronic patient record system (e.g. medication modules) would likely qualify in their own right as medical devices. The difficulty of the previous description highlights the need for healthcare organisations and authorities to raise clinicians’ awareness and understanding of reportable EHR issues, which would contribute to the reporting of incidents and improve the regulatory system’s effectiveness.
The broader picture of EHR safety needs to be discussed in this context because also current overlapping systems, such as voluntary patient safety incident reporting used for handling EHR safety issues, is perhaps not the most effective way to manage with these complicated safety issues.35,36 When clinicians must use multiple reporting channels, the number and quality of reporting inevitably comes into question. The purpose and processes of multiple EHR safety reporting systems must be clarified, and overlapping systems should be harmonised to make them more effective.
To maximise learning, lessons learnt from incidents, descriptions of implemented risk controls and their effectiveness should be shared within and between organisations. 37 Similar events will most likely occur at other institutions, 5 and the data could provide lessons worth learning. Every single case in this article highlights an EHR vulnerability that healthcare organisations should be aware of in order to ensure that the error does not occur in their own environment. A regulatory database for sharing experiences of medical software errors with the goal of developing prevention strategies 2 does not exist in Finland. Thus, we suggest considering an establishment of this kind of database.
A classification system that distinguishes error types is necessary to more effectively use these data. 18 The use of a data structure is also important in helping clinicians understand the various types of incidents that can occur with HIT. We therefore recommend the use of certain classifications so as to facilitate analysis and learning. The FIN-TIERA tool based on research of commonly recognised error types was applied in this study. 25 During this study, a new error type of patient data privacy was detected, which suggests further research in this important issue a new error type of data privacy supported by the literature. 38 An error type of data privacy will be added for a next test phase of the tool.
Limitations
One limitation of this study is the relatively small number of reports for analysis. Only 138 EHR user cases were reported to the supervisory authority during the 5-year period. One obvious reason for this low number is the underreporting of adverse events, due in part to clinicians’ lack of awareness of the reporting criteria described in the previous section. Moreover, the vendors’ completion of the conformity marking (CE) application process affected the selection criteria of this study. However, despite our small sample size, our in-depth analysis, including a blinded inter-rater review, presents a valuable picture of medical software user reporting, as the analysis revealed a significant difference in the reporting rate compared to different data sources.
Conclusion
This analysis provides data from a setting with a 100 per cent EHR implementation rate and lots of experience with EHR use. Adverse events associated with EHR vulnerabilities in this study, which led to serious harm, occur in all types of healthcare settings and cluster around certain error types; these data could prove useful in surveillance efforts to reduce the risk of patient harm in the future. We recommend classifying incidents according to a common taxonomy. The low number of reported incidents raises questions not only about the challenges associated with medical software oversight but also still existing barriers for incident reporting.
Footnotes
Acknowledgements
The authors thank the Finnish National Supervisory Authority for Welfare and Health (Valvira) for their assistance with the data collection.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work benefited from the support of Finnish governmental research funding (study grant TYH2014224).
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.