
Abstract
This study evaluates the performance of a set of machine learning techniques in predicting the prognosis of Hodgkin’s lymphoma using clinical factors and gene expression data. Analysed samples from 130 Hodgkin’s lymphoma patients included a small set of clinical variables and more than 54,000 gene features. Machine learning classifiers included three black-box algorithms (k-nearest neighbour, Artificial Neural Network, and Support Vector Machine) and two methods based on intelligible rules (Decision Tree and the innovative Logic Learning Machine method). Support Vector Machine clearly outperformed any of the other methods. Among the two rule-based algorithms, Logic Learning Machine performed better and identified a set of simple intelligible rules based on a combination of clinical variables and gene expressions. Decision Tree identified a non-coding gene (XIST) involved in the early phases of X chromosome inactivation that was overexpressed in females and in non-relapsed patients. XIST expression might be responsible for the better prognosis of female Hodgkin’s lymphoma patients.
Keywords
Introduction
Hodgkin’s lymphoma (HL) is a haematological malignancy accounting for about 10 per cent of all lymphoma cases in Western countries.1,2 HL is composed of two distinct disease entities: classical HL, which accounts for about 95 per cent of the whole disease burden and is characterized by the presence of malignant multinucleated giant Reed–Sternberg cells, and nodular lymphocyte predominant HL, characterized by a neoplastic population of larger cells with folded lobulated nuclei. 3
In the last decades, advances in radiation treatments and chemotherapy have greatly increased the survival rates of HL patients. Nonetheless, up to date, about 5–10 per cent of them are refractory to initial treatment and 10–30 per cent will relapse despite having achieved an initial complete remission. 4
IPS (International Prognostic Score) is a prognostic index based on the combination of seven recognized prognostic factors for HL (namely, age ⩾45 years, stage IV, male sex, white blood count ⩾15,000 cells/mL, lymphocyte count <600 cells/mL, albumin < 4.0 g/dL, haemoglobin < 10.5 g/dL). 5 IPS was demonstrated to be predictive of the patient outcome in multivariable analysis. For instance, patients with five or more factors were found to have a 5-year progression-free survival of 42 per cent, while patients with non-negative prognostic factors had an 84 per cent probability of being free from progression at 5 years from diagnosis. 5 However, despite the quite good performance of IPS, the identification of new prognostic variables for HL patients is highly desirable to potentially increase patient survival and reduce treatment toxicity. 4 For this purpose, numerous studies have been carried out in the last few years and many new putative prognostic markers have been identified.6,7 Among such studies, a large microarray experiment identified a set of 271 genes differently expressed between relapsed and non-relapsed patients. 8 Furthermore, the same study was able to associate a macrophage gene expression signature with primary treatment failure, even if this latter finding was questioned by further investigations.9,10
This study is aimed at evaluating the performance of a set of supervised machine learning techniques, including the recently proposed Logic Learning Machine (LLM) method, in predicting the prognosis of HL patients using clinical and gene expression data from the large data set by Steidl et al. 8
Materials and methods
Database description
Data were downloaded from GDS4222.soft, a microarray database stored in the GEO repository 11 at http://www.ncbi.nlm.nih.gov/sites/GDSbrowser?acc=GDS4222 . Data included information from 130 samples of classical HL and 54,675 gene expression features. 8
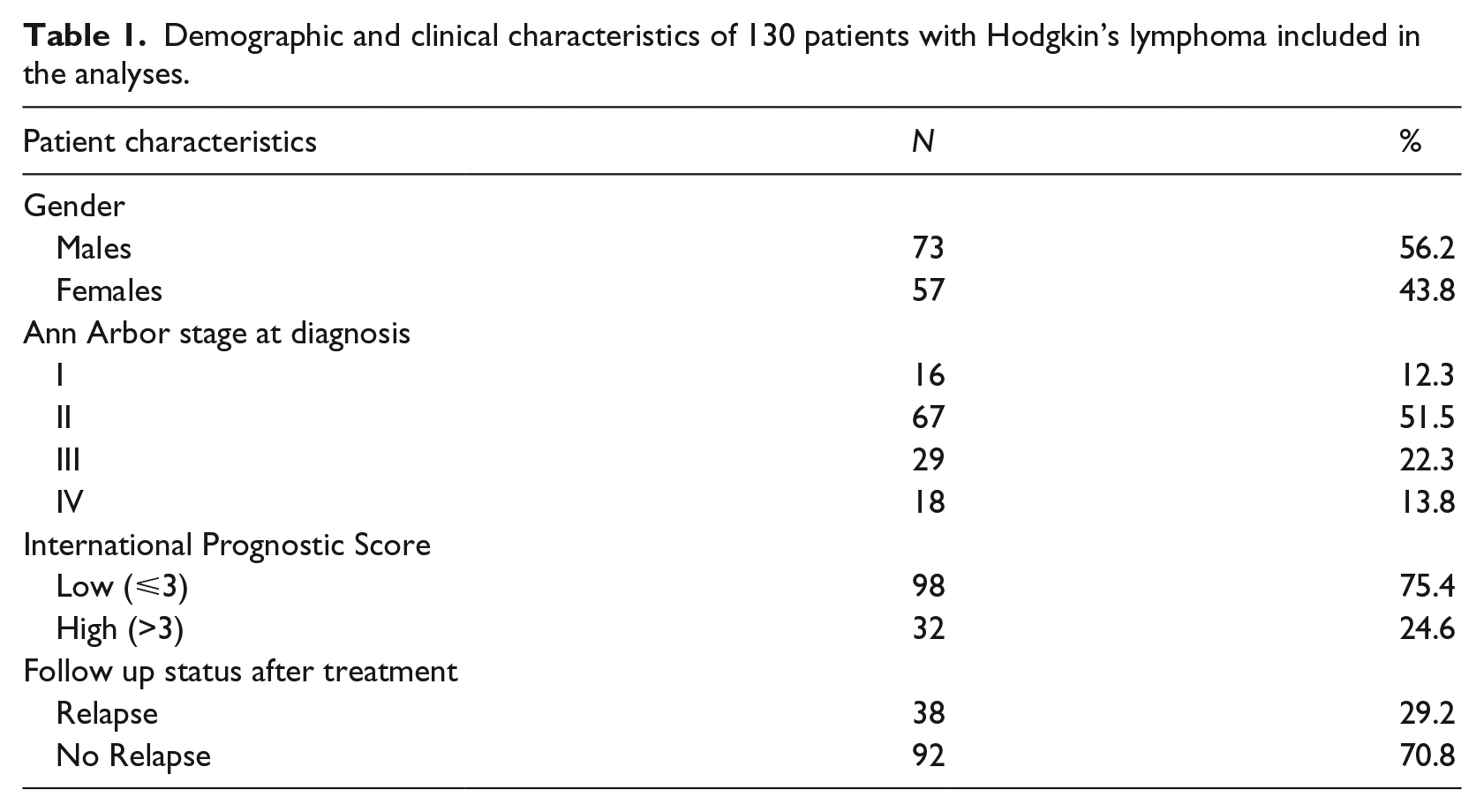
Table 1 describes patient characteristics available in GDS4222.soft. Clinical and demographic variables included the following: relapse at any time after therapy (n = 38, 29.2%), gender (male: 56.2%; female: 43.8%), stage at diagnosis (Stage I: 12.3%; Stage II: 51.5%; Stage III: 22.3%; Stage IV: 13.8%) and IPS. This latter was aggregated into two categories, according to Steidl et al. 8 : high score, corresponding to IPS > 3 (24.6%), and low score, associated with IPS = 3 (75.4%). More details about patient selection, characteristics at diagnosis, assessment of disease status, primary line treatment, and methods for gene expression analysis, including data pre-processing and normalization, have been reported elsewhere. 8
Demographic and clinical characteristics of 130 patients with Hodgkin’s lymphoma included in the analyses.
Supervised data mining methods
A set of supervised learning machine techniques was selected in order to predict HL patient prognosis. They included three common methods based on black-box algorithms (k-nearest neighbour classifier, kNN; Artificial Neural Network, ANN; and Support Vector Machine, SVM) and two methods based on intelligible threshold-based rules (Decision Tree, DT, and the innovative LLM method). Standard classification based on the IPS score was also performed. The probability of relapse at any time after therapy was considered as the outcome, while all the available variables were used as input data. Accuracy measures included the total proportion of correctly classified samples (total accuracy) and the proportion of correct classifications among both relapsed (sensitivity) and non-relapsed patients (specificity).
In order to control the overfitting bias, accuracy estimates of each supervised analysis were obtained by cross-validation. Due to the rather small sample size of our data set, the leave-one-out procedure was adopted. 12 Finally, a comparison between the set of intelligible rules generated by LLM and DT was also performed.
All the analyses were carried out by using Rulex Analytics, a software suite developed and commercialized by Rulex Inc ( http://www.rulex-inc.com ).
kNN
Consider a training set S including n input–output pairs (
In the present investigation, the set of values {1, 3, 5} was adopted for k and the standard Euclidean distance was employed, after having normalized the components of the input vector
ANN
ANN is a connectionist model formed by the interconnection of simple units, called neurons, arranged in layers. The first layer receives the input vector
In this study, one intermediate layer was used, and the number of hidden neurons was allowed to vary from one to three. The nets were trained by means of the Levenberg–Marquardt version of the back propagation algorithm. 13
SVM
SVM is a non-probabilistic binary linear classifier based on the identification of an optimal hyperplane of separation between two classes. Given a training set, the classifier selects a subset l of input vectors
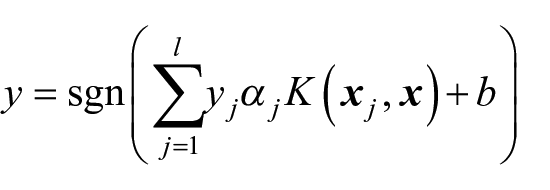
where the coefficients α j and the offset b are evaluated by the training algorithm.
K(·,·) is a kernel function used to perform a non-linear classification by constructing an optimal hyperplane in a high dimensional projected space. Both a linear and a radial basis kernel function were tested on the GDS4222.soft data set. As it will be shown in the following section, in this case the linear kernel (which produces a linear classification) proves to be more robust with respect to overfitting. This is due to the fact that the classification problem is unbalanced (38 patients relapsed, while 92 did not), and moreover, the number of input attributes for classification far exceeds the number of training samples. The training algorithm was performed using the LIBSVM library, which is featured by the Rulex Analytics software.
DT
A DT is a graph where each node is associated with a condition based on an attribute of the input vector
In the present investigation, the information gain IG (also called ‘the smallest maximum entropy’) was employed as goodness indicator function. In more detail, given a set Q and a partition in q subsets Q1,…, Qq, the information gain of Q with respect to the partition
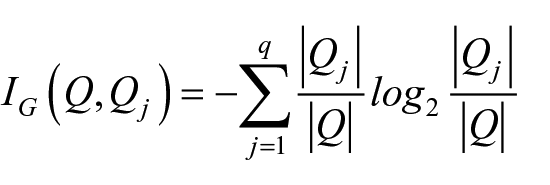
where | . | indicates the number of elements in a set.
In our study, q = 1 identifies the subset of non-relapsed patients and q = 2 the subset of relapsed ones.
Finally, the pessimistic error pruning technique was adopted to reduce the complexity of the final DT and to increase its generalization ability. Briefly, let p be the error rate associated with a node s in a DT; all nodes and leafs below s are erased if the error p–s associated with the node immediately below s exceeds the following quantity
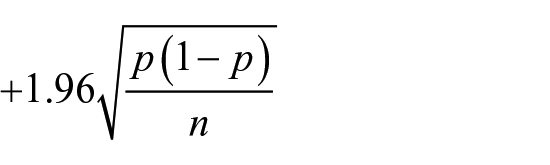
where n represents the number of samples to be classified at the node r. 14
LLM
LLM is an innovative method of supervised analysis based on an efficient implementation of the Switching Neural Network model,15,16 which is associated with a classifier g(
The general procedure employed to train an LLM passes through the following steps:
Discretization. Continuous and integer variables are properly discretized to reduce their variability, thus increasing the efficiency of the training algorithm and the accuracy of the resulting set of rules.
Binarization. Nominal and (discretized) ordered variables are coded into binary strings by adopting a suitable mapping that preserves ordering and distances.
Logic synthesis. Starting from the binarized version of the training set S, which can be viewed as a portion of a truth table, reconstruct the
Rule generation. Transform every logical product of the
A valid and efficient way of performing Step 1 consists in adopting the attribute-driven incremental discretization (ADID),17,18 which reduces the complexity of the input vector
Then, the (inverse) only-one coding
15
is adopted at Step 2 to transform the training set S into a collection of binary strings that can be viewed as a portion of the truth table of a monotone Boolean function. Here, for each (binarized version of a) pattern
To ensure a good generalization ability, the logic synthesis (Step 3) is performed via an optimized version of the Shadow Clustering (SC) algorithm, 16 a proper technique for reconstructing monotone Boolean functions starting from a partially defined truth table. In contrast with methods based on a divide-and-conquer approach, SC adopts an aggregative policy, that is, at any iteration some patterns (coded in binary form) belonging to the same output class are clustered to produce an intelligible rule. A suitable heuristic approach is employed to generate implicants (rules) exhibiting the highest covering and the lowest error; a trade-off between these two different objectives generally leads to final models showing a good accuracy.
The training algorithm for LLM requires to define a single parameter ϵ, the maximum error that can be scored by each generated rule. In all our trials, we have used the value ϵ = 0.
Results
Table 2 resumes the performance of standard clinical classification in leave-one-out cross-validation, based on the IPS index, and that of the selected supervised methods. IPS correctly classified 68 per cent of total patients, with 37 per cent sensitivity and 80 per cent specificity.
Comparison between standard clinical classification by IPS score and the selected methods of supervised analysis in leave-one-out cross-validation.
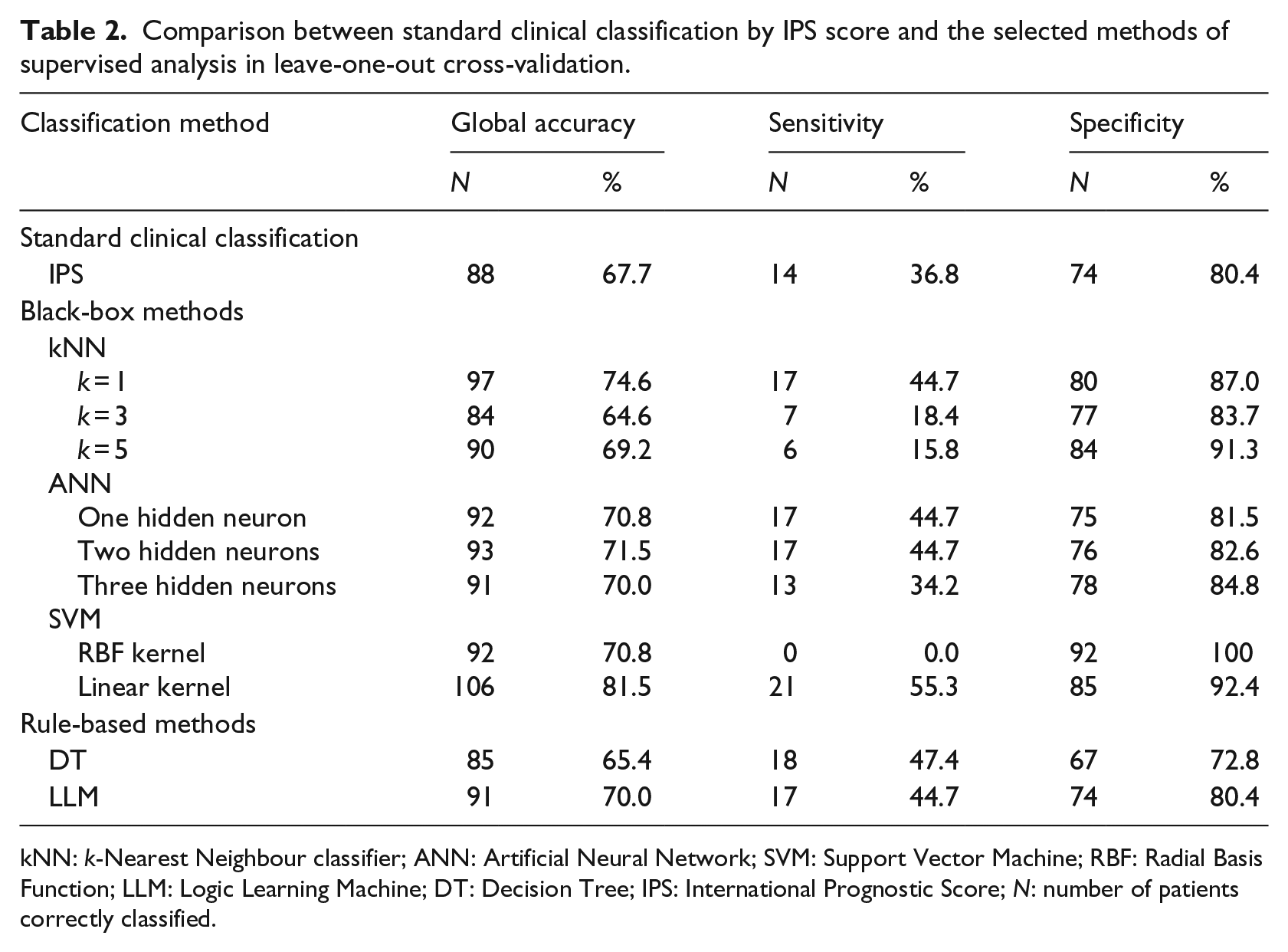
kNN: k-Nearest Neighbour classifier; ANN: Artificial Neural Network; SVM: Support Vector Machine; RBF: Radial Basis Function; LLM: Logic Learning Machine; DT: Decision Tree; IPS: International Prognostic Score; N: number of patients correctly classified.
Among the three black-box methods, the best performance was achieved by SVM with linear kernel (global accuracy = 82%, sensitivity =55%, specificity = 92%). kNN with k = 1 also outperformed the standard clinical classification (global accuracy = 75%, sensitivity = 45%, specificity = 87%), whereas models with higher k values showed a poor performance and, in particular, a very low sensitivity. With regard to ANN, the model with two hidden neurons shows the highest performance, which lies between that of IPS only and that of kNN (global accuracy = 72%, sensitivity = 45%, specificity= 83%).
Among the two considered rule-based methods, LLM showed the best performance (global accuracy =70% vs 65% for DT), even if sensitivity was slightly lower (45% vs. 47%).
When the analysis was repeated on the whole data set, LLM selected 25 rules that included a minimum of two and a maximum of six conditions; the corresponding covering ranged between 2.2 and 53.3 per cent.
Table 3 shows the rules generated by LLM after the exclusion of those with a low coverage (<20%). This restriction was made in order to reduce the effect of outliers, thus allowing a more reliable comparison with DT after the pruning procedure.
Classification rules identified by the Logic Learning Machine on the whole data set.
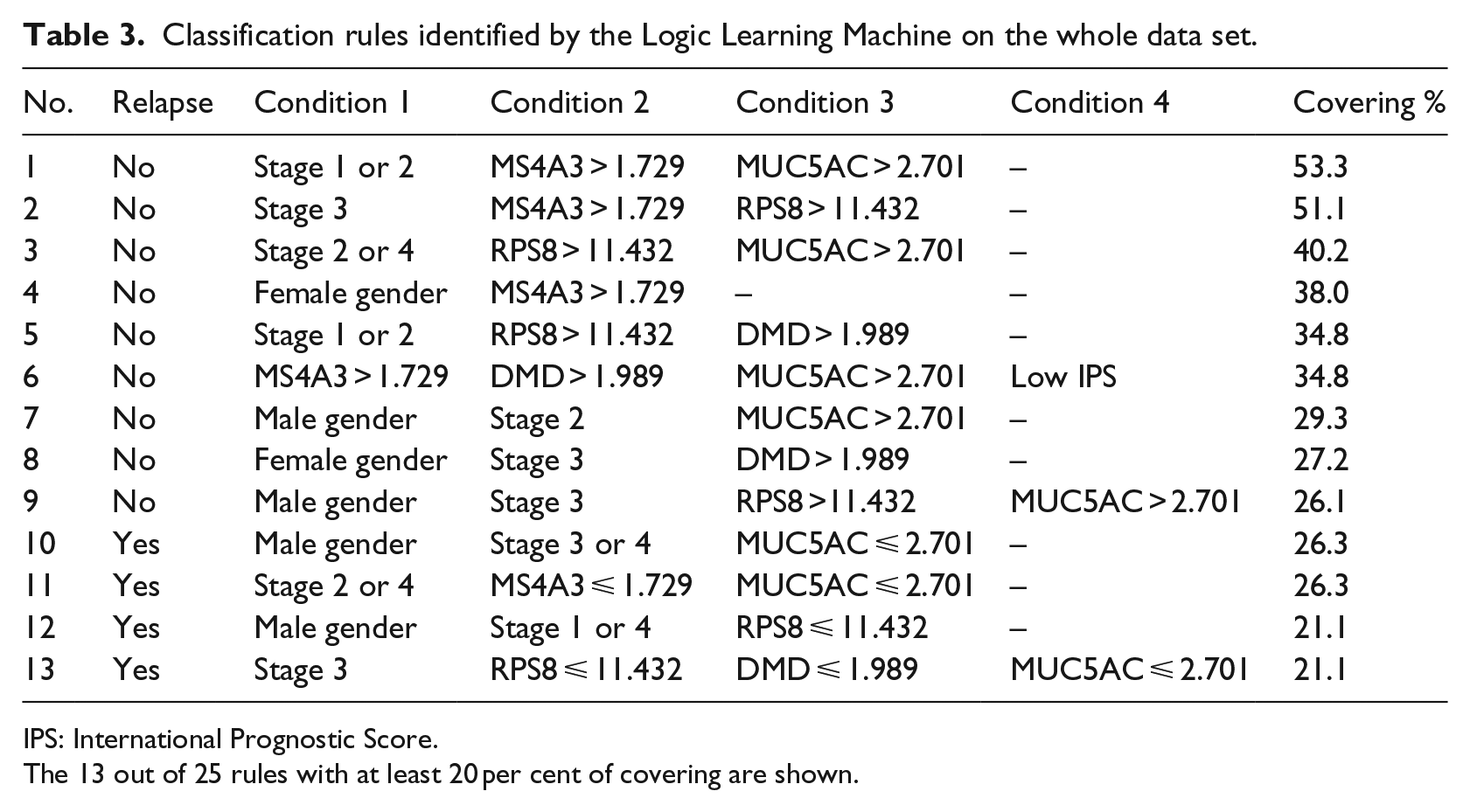
IPS: International Prognostic Score.
The 13 out of 25 rules with at least 20 per cent of covering are shown.
All the LLM rules included at least one clinical or demographic characteristic of patients. On the whole, LLM identified four features relevant for classification, all inversely associated with the occurrence of relapse (namely, MS4A3, RPS8, DMD and MUC5AC). With regard to clinical conditions, advanced stages (3 and 4) were more often associated with relapse, but with some exceptions (e.g. rule 2, condition 1). IPS was included in only one rule (no. 6), and as expected, a low value corresponded to the absence of relapse. Finally, gender was included in 6 out of 13 rules. Among the four rules identifying relapsed patients, two included males (no. 10 and no. 12, respectively, condition 1), whereas females were never selected.
Figure 1 shows the classifier obtained by DT. Classification was performed by seven rules that involved gene expression only (namely, XIST, EPOR, GPR82, AV719529 and KIAA1430). A prediction of relapse was associated with low values of XIST and GPR82 and high values of AV19529 and KIAA1430.
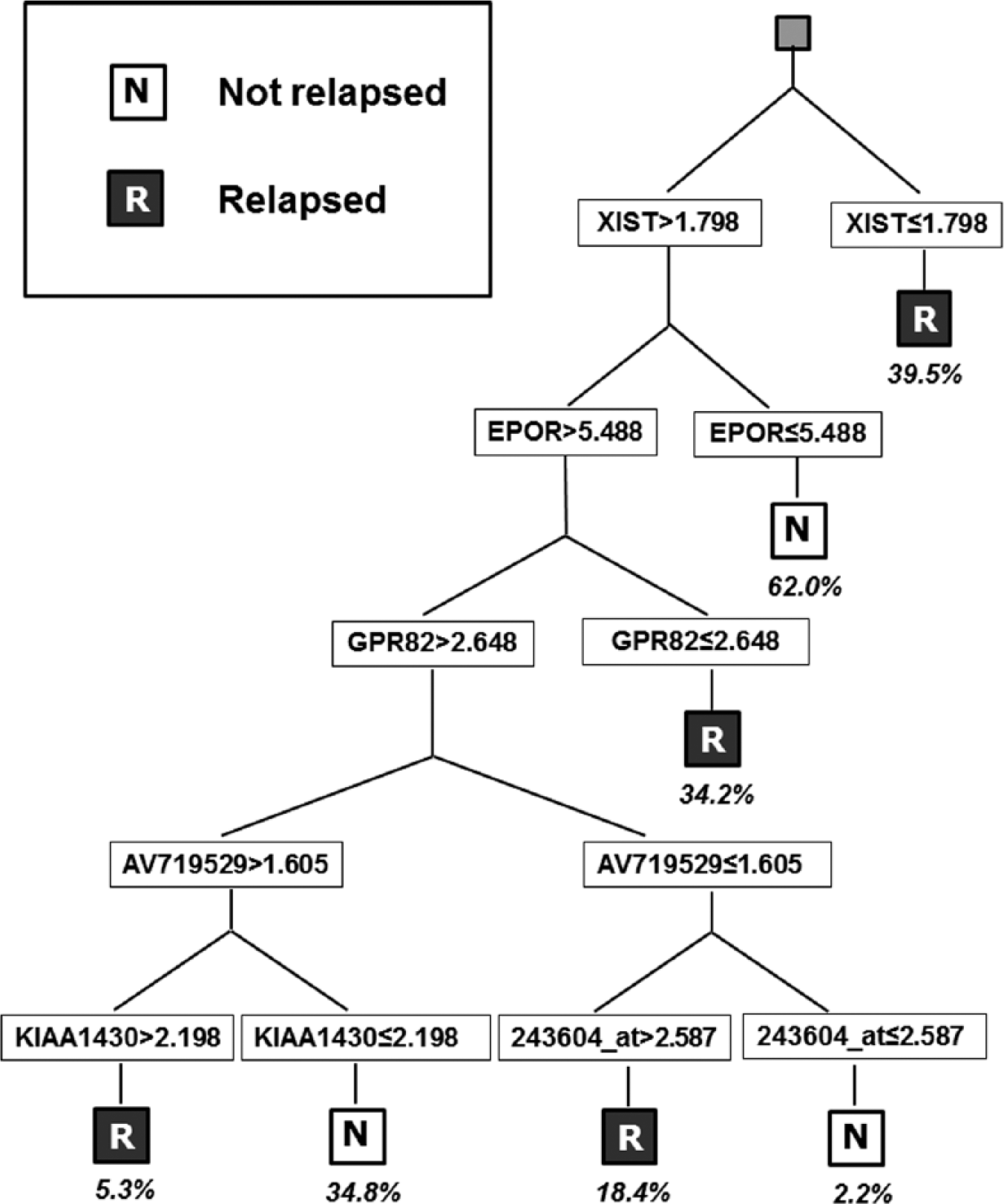
Classification obtained by DT on the whole data set. Percentages indicate the covering of each rule.
Discussion
Despite advances in therapeutic treatment, about 20 per cent of HL patients eventually die, whereas a similar proportion is likely to be over-treated. 8 The large availability of new potential tumour markers for HL prognosis, including genome-wide gene expression data, might contribute to the improvement of the performance of IPS in predicting patient survival.7,8,19
Many supervised methods of data analysis are available to exploit and combine information from new tumour markers and clinical prognostic factors. In particular, ANN, kNN and the more recent SVM have shown a high accuracy in predicting survival of cancer patients when applied to gene expression data in many different clinical settings.20–26 However, such algorithms are usually referred to as ‘black-box’ methods since classification is made through a mathematical formula that makes it difficult to evaluate the biological and clinical role of variables included in the analysis. Conversely, algorithms based on intelligible threshold rules, like DT and the recently proposed LLM, can provide useful information for a better understanding of tumour biology and for addressing therapeutic approaches.18,23
The good performance of LLM compared to that of common supervised techniques was demonstrated in a set of biomedical studies.18,27,28 However, different from DT,20–29 LLM has been never applied for classification purposes to large databases of highly correlated features, such as microarray gene expression data. In this study, in agreement with results from previous investigations, LLM showed a performance quite similar to that of some common competing black-box methods (ANN and kNN), but lower than that of SVM.
LLM outperformed DT and was able to combine information from clinical variables with expression values from a small panel of selected genes. In particular, stage and gender were in some cases associated in the same rule, but never associated with IPS (Table 2). Since IPS is constructed using clinical variables that also include stage and gender, 5 this finding suggests that LLM tends to reject redundant information. Furthermore, a low IPS score, a low stage at diagnosis and female gender were more often associated with a good prognosis, in agreement with knowledge from previous investigations. 5
Taken together, these results suggest that the combination of clinical data and gene expression features could provide useful information for assessing the prognosis of HL patients. This observation is in agreement with previous studies on different malignancies, indicating that clinical information can enrich microarray data in identifying a suitable classifier for the prediction of cancer survivability.23,30,31
Gene expressions selected by LLM were all different from those identified by DT, and they also differed from the 30 most relevant features identified by the original analysis. However, MUC5AC and EPOR were also included into the complete list of differentially expressed genes reported by Steidl et al. 8 The four genes identified by LLM were all under-expressed in relapsed patients. MS4A3 (membrane-spanning 4-domains subfamily A member 3) is localized in 11q12 and encodes a membrane protein probably involved in signal transduction. 32 Interestingly, MS4A3 belongs to the same membrane-spanning 4-domains gene subfamily of MS4A4, which was recognized to be associated with HL prognosis in previous investigations. 33 RPS8 is localized in 1p34.1-p32 and encodes a ribosomal protein that is a component of the 40S subunit. 34 DMD (dystrophin) locates at Xp21.2 and is a highly complex gene, containing at least eight independent, tissue-specific promoters and two polyA-addition sites. 35 Finally, MUC5AC is located in 11p15.5 36 and encodes for a protein (mucin) involved in secretion of gastrointestinal mucosa.
With regard to DT, genes with a known function ( http://www.ncbi.nlm.nih.gov/gene ) include the following: XIST (X inactive specific transcript), which is a non-coding gene located in Xq13.2, involved in the inactivation of X chromosome in human females, 37 and EPOR, located in 19p13.3-p13.2, which encodes an erythropoietin receptor. 38 Moreover, GPR82, localized in Xp11.4, encodes for a protein with unknown function but is a member of a family of proteins that contain seven transmembrane domains and transduce extracellular signals through heterotrimeric G proteins. 39 Interestingly, partly consistently with our observation of a higher relapse probability among subjects with low XIST expression, XIST was demonstrated to activate apoptosis in T lymphoma cells via ectopic inactivation of the X chromosome. 40 In our data, XIST was strongly overexpressed among females (data not shown), thus potentially providing a new insight about the biological mechanism at the basis of the better prognosis commonly observed among females.
Results of our study may be prone to some limitations. In particular, we selected the GDS4222 data set because, at least to our knowledge, it was among the biggest publicly available gene expression databases including information about prognosis of HL patients. However, as a whole, its sample size (130 patients, including 38 relapsed) was too small to allow drawing definitive conclusions, and all findings reported in our study need confirmation by other independent investigations. Furthermore, sensitivity of any applied method (including SVM) was unsatisfactory (<60%). In fact, in the presence of unbalanced outcomes, as in our study, rules extracted from LLM can be weighted to improve their accuracy. 17 According to this property, we tried to reclassify a posteriori the patients under study by assigning a 1:10,000 weight in favour of relapsed outcome, but also in this further analysis sensitivity never achieved 60 per cent (data not shown), pointing out that the limit of 60 per cent for sensitivity is difficult to be exceeded for any of the considered methods. The lack of potentially relevant clinical information (e.g. absolute lymphocyte count, age at diagnosis and first line treatment) and the poor measure of the outcome, which did not include time-to-event values, could have contributed to lowering the sensitivity of our study. Moreover, we performed all the analyses without applying any pre-filtering technique to the data under study. Previous investigations have demonstrated that the performance of supervised methods can be enhanced by applying pre-filtering and feature selection methods, which can reduce overfitting.41–43 Their effect on LLM classification has not been investigated yet.
Conclusion
LLM provided simple intelligible rules that could contribute to the knowledge of HL biology and to address therapeutic approaches by combining clinical information and gene expression data.
The role of genes identified by both LLM and DT in the clinical course of HL patients should be investigated in further studies. In particular, the higher expression of XIST in patients with a good outcome and among females might be related to the still unknown factors favouring the better prognosis of female patients with HL.
Footnotes
Acknowledgements
Stefano Parodi is a research fellow of the Italian MIUR Flagship project ‘InterOmics’.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship and/or publication of this article.