
Abstract
In the United Kingdom, there are more than 9000 reports of adverse events involving medical devices annually. The regulatory processes in Europe and in the United States have been challenged as to their ability to protect patients effectively from unreasonable risk and harm. Two of the major shortcomings of current practice include the lack of transparency in the safety certification process and the lack of involvement of service providers. We reviewed recent international standardisation activities in this area, and we reviewed regulatory practices in other safety-critical industries. The review showed that the use of safety cases is an accepted practice in UK safety-critical industries, but at present, there is little awareness of this concept in health care. Safety cases have the potential to provide greater transparency and confidence in safety certification and to act as a communication tool between manufacturers, service providers, regulators and patients.
Introduction
Recent debates in the The Lancet and British Medical Journal (BMJ) highlighted serious concerns about the regulation of medical devices in Europe.1,2 Medical devices are implicated in a significant number of adverse events. For example, during 2005–2009, more than 56,000 adverse events and 710 deaths involving infusion devices were reported to the US Food and Drug Administration (FDA). 3 In the United Kingdom, the Medicines and Healthcare Products Regulatory Agency (MHRA) received 9099 reports of adverse events involving medical devices during 2009, 4 including 1885 cases of serious injury and 202 deaths. 5 In a BMJ feature, Cohen describes the case of the articular surface replacement (ASR) hip implant that had been used on 93,000 patients before its withdrawal from the market in 2010 following a large number of adverse events involving the device. 6 Metal debris from the implant had destroyed the soft tissue surrounding the joint and also affected the bones in some patients. The device had entered the European market in 2003 without clinical studies. By 2007, surgeons were reporting increased anomalies associated with implanted devices, but the manufacturer put down such reports to deficient surgical technique. The MHRA required further 3 years to issue a device alert in 2010, and later that year, the manufacturer voluntarily phased out the ASR devices due to commercial performance reasons, stating that this decision had not been related to any safety concerns. Cohen 6 concludes that this and similar cases demonstrate ‘the power that companies have in deciding the fate of their devices, their hold over surgeons, and the lack of regulatory power in Europe’.
Particular criticism of the role of the MHRA has been brought forward by the editor of The Lancet following the recent investigation into adverse events associated with Poly Implant Prostheses (PIP) breast implants. 7 The manufacturer had used a non-approved low-grade silicon gel. This led to an increased risk of rupture and leakage of the approximately 240,000 implants that 130,000 women received in the United Kingdom during 2001–2011. The final report by the UK Department of Health expert group published in June 2012 concludes that the providers of PIP implants should contact women to offer consultation and removal of the implant where appropriate. 8 The recent editorial in The Lancet challenges the role and efficacy of the MHRA in protecting the public from harm, suggesting that ‘these serious examples of device failures result from the MHRA’s paralysis and inability to address the shortcomings of a badly flawed system’. 1 In a response statement, the MHRA denies such allegations but adds that recommendations have been made to the European Commission including ‘proposals to improve oversight of Notified Bodies, the surveillance of post-market events and the collaboration between national regulatory bodies’. 9
One implication of the current regulatory framework is that device manufacturers are responsible for determining acceptable levels of risk,10 –12 in particular in the European market where certain classes of devices undergo a self-certification process or are certified based on evidence of an appropriate quality assurance system. 13 For programmable electrical medical systems (PEMS), manufacturers are responsible for ensuring that the device is adequately safe for use in a specific context. However, the manufacturer usually has limited control over how devices are used in the operational context, and whether critical assumptions about aspects such as training and maintenance are fulfilled. The health-care service provider often has to integrate a number of different devices and other health information technology (IT) products within their environment. 14 Without guidance and appropriate standards, devices may not work properly when integrated into a service provider’s network or they may interact in unexpected ways with other devices and IT products.14,15 The safety of the resulting system can only be assured if the service providers are involved appropriately, and if evidence provided by both the manufacturer and the service provider are accessible and integrated adequately.
This lack of adequate communication between stakeholders and the unavailability of device-related data have been recognised specifically for networks incorporating medical devices and generic health IT software products, and international standards have been developed to overcome some of the limitations. In this article, we review some of the relevant standardisation efforts by the International Electrotechnical Commission (IEC), the International Organization for Standardization (ISO) and the UK National Health Service (NHS). In a review of regulatory practices in safety-critical industries in the United Kingdom, 16 we identified the use of safety cases as an accepted best practice. In health care, the use of safety cases could serve as a communication tool between manufacturers, service providers, the regulator and the public, and it could encourage greater transparency and confidence in the safety assurance of medical devices and health IT products.
The next describes the regulatory context, current risk management practice and recent standardisation efforts in the medical device area. In the subsequent section we outline a simple high-level safety argument over the life cycle of a generic medical device or health IT product as a potential way of involving health-care organisations in the assurance of safety. We conclude the paper with a discussion of opportunities and challenges for the development and use of safety cases in health care.
Regulatory context and current practices
In the regulation of health-care systems, there is a differentiation between manufacturers of medical devices and health IT products on the one hand, and health-care providers as users or consumers of such products on the other hand. In general, manufacturers of medical devices have to provide evidence that their devices are acceptably safe for a particular use in a specific environment. Health-care providers, on the other hand, are audited to ensure that the care they provide meets national standards. A part of this is the requirement to use only previously certified medical devices.
Regulation addressed to medical device manufacturers
The definition of what constitutes a medical device is broad and comprises devices as diverse as radiation therapy machines, infusion pumps and wheelchairs. The European Medical Devices Directive (MDD) 17 specifies essential requirements that have to be met by any device to be marketed in the European Union (EU). It provides classification rules based on certain characteristics of a medical device, as well as conformity routes that specify different ways of manufacturer compliance with the essential requirements based on the class of the medical device under consideration. Compliance with the essential requirements is demonstrated through display of the Conformité Européenne (CE) marking. The assessment of compliance is undertaken by notified bodies rather than a central regulatory authority. Notified bodies are commercial organisations, certified by national regulators, who perform compliance assessment as clients of the device manufacturer. At present, there are 74 notified bodies in the EU. 18 This implies that assessments may vary depending on the notified body carrying out the assessment18,19 and that the data submitted as part of the compliance assessment (including product description, literature reviews, risk analysis results and testing and inspection reports) are protected by confidentiality agreements between the parties and are hence unavailable to clinicians or the public. For medical devices belonging to Class I (typically devices perceived as posing lower levels of risk), the manufacturer can claim compliance through a self-certification process.
The approach to regulation practised in the United States through the FDA is different from the European approach. There are essentially two routes to pre-market approval: the Premarket Authorisation (PMA) process involving more stringent requirements for new devices and the 510(k) process, which is an abbreviated approval process for devices that can claim substantial equivalence with devices already on the market. While the FDA approach is regarded by some as better suited to ensure clinical effectiveness of devices and patient safety, 18 a recent study investigating 113 recalled devices that had caused serious health problems found that most had been approved through the 510(k) route or had been deemed such low risk that they were exempted from regulatory review. 20
International standards
In Europe, more than 200 standards relating to safety of medical devices are harmonised and provide a technical interpretation of the essential requirements of the MDD. 17 The main safety standards for electrical medical systems are the standards of the IEC 60601 series. The IEC 60601-1 series of standards consists of ‘Part 1: General Requirements for Basic Safety and Essential Performance’, 11 and a number of collateral standards addressing specific requirements for particular systems. Since 2005, the third edition of IEC 60601-1 includes an update of the requirements of the second edition as well as new solutions now possible due to the availability of novel technology. The most significant change is the introduction of the notion of risk management by integration of standard ISO 14971, 12 see later in the article, in order to make the standard more flexible with respect to the rapid growth in technology by allowing manufacturers greater freedom of how they mitigate safety threats. For manufacturers, this provides greater flexibility, but at the same time, it requires that manufacturers determine risk acceptability based on a risk management process compliant with ISO 14971. The transition dates from the second edition to the third edition are June 2012 for Europe and June 2013 for the United States, respectively. Figure 1 provides an overview of the evolution of IEC 60601-1.
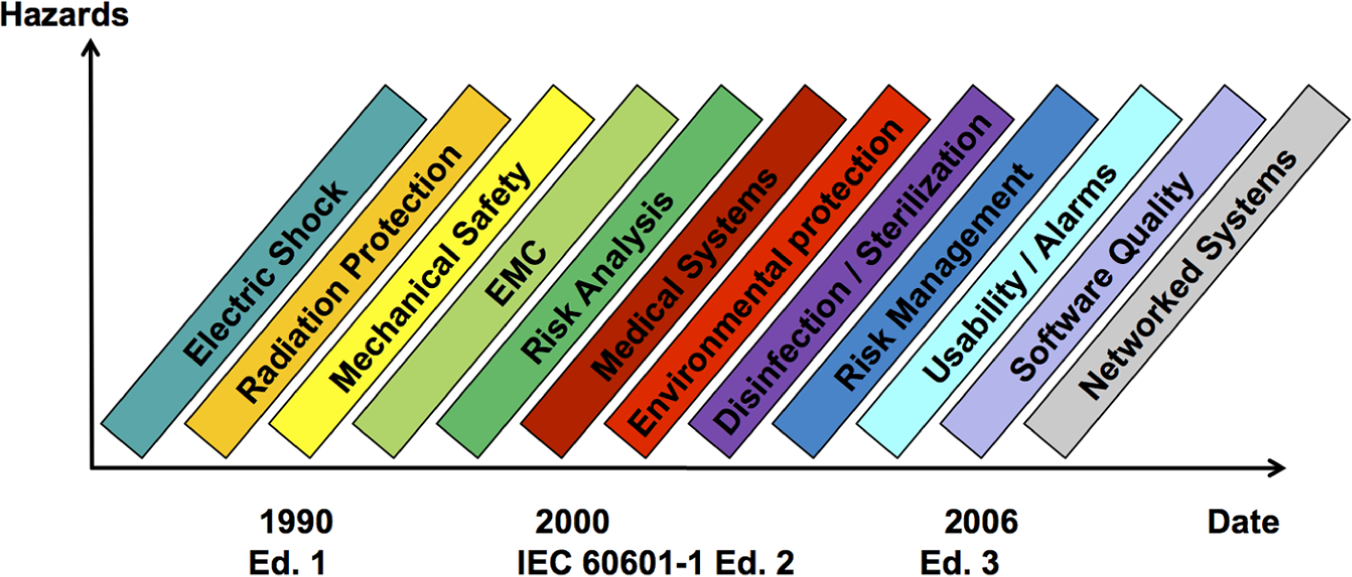
Evolution of IEC 60601.
Risk management
A risk management process for the manufacture of medical devices is specified in ISO 14971 – ‘Application of Risk Management to Medical Devices’, now in its second edition.
12
This industry standard requires that the manufacturer shall establish, document and maintain throughout the life cycle, a process for identifying hazards associated with a medical device, estimating and evaluating the associated risks, controlling these risks and monitoring the effectiveness of the controls. The manufacturer needs to demonstrate that the residual risk of using the medical device is acceptable. In Section D5.5, the standard links back to standards such as IEC 60601 in order to simplify, where appropriate, the task of analysing the remaining residual risk:
When relevant safety standards exist, they can address some or all of the risks that need to be dealt with for a particular medical device. It is presumed that, in the absence of objective evidence to the contrary, meeting the requirements of the relevant standards results in particular risks being reduced to an acceptable level, but the responsibility for verifying that this is the case for a particular device rests with the manufacturer.
12
Compliance with the performance of the risk management process is done by inspection of the risk management file (RMF). The RMF is a repository containing the set of records produced by the risk management process, and remains the manufacturer’s property that is not available to users or the public. The RMF is not intended to be a structured argument that the device is acceptably safe for use but acts as a document repository that provides traceability about the various hazards that have been considered. Inspection of the RMF focuses on the quality of the process, rather than actual results (e.g. of the risk analysis), and the acceptability of risk is determined by the manufacturer’s policy for risk acceptability.10–13 ISO 14971 states that this policy should consider known stakeholder concerns and the currently accepted state of the art. The regulator can refuse certification if the risk management process is perceived to be inadequate. In Europe, notified bodies, whose role has been criticised recently for the lack of transparency and consistency, conduct conformity assessment, and calls for an independent government agency (similar to the FDA in the United States) have been put forward.5,18
IEC 80001 – risk management for IT networks incorporating medical devices
Device manufacturers and bodies such as the FDA have recognised the challenges posed by networked and IT-based health-care systems and have been engaged in relevant standardisation efforts for the past 5 years.
IEC 80001-1 14 entitled Application of Risk Management for IT-Networks Incorporating Medical Devices – Part 1: Roles, Responsibilities and Activities defines roles and responsibilities, and describes a risk management process in order to ensure interoperability of medical devices connected through IT networks without compromising the organisation and delivery of health care in terms of safety, effectiveness and data and system security.
The standard identifies the service provider as responsible organisation with which overall responsibility for the risk management process of IT networks incorporating medical devices rests. The responsible organisation needs to appoint an IT-network risk manager who owns and implements the risk management process. The standard recognises the information needs of the responsible organisation and defines responsibilities for manufacturers to supply relevant information in the accompanying documents.
While the risk management process is based on the familiar steps defined in ISO 14971, this standard represents a major change in thinking in as far as it recognises explicitly the need to assess safety from an operational perspective throughout the life cycle of medical devices. Ultimate responsibility for determining acceptability of risk is with the service provider who, on the one hand, needs to implement an appropriate risk management process, but who, on the other hand, also has the right to expect suitable communication with the manufacturer and disclosure of safety-related information and assumptions.
Lack of involvement of health-care service providers
Until recently, the whole set of standards on safety of medical systems put the manufacturer in the position of the decision-maker of risk acceptance, even though the regulator can refuse certification in those cases where the manufacturer’s processes are perceived to be lacking in quality. The underlying assumption is that the manufacturer defines normal use, and that all hazards, associated risks and acceptance criteria regarding a particular medical system have been elicited with adequate resources using appropriate clinical information and expertise, and that they have been recorded in the RMF. In Europe, this information is not available to clinicians, patients or the public due to commercial confidentiality reasons.
Apart from issuing instructions for use, the manufacturer of common medical devices has little influence on the way the devices are actually used in practice. This is particularly problematic in situations where the acceptability of the overall residual risk may depend on risk controls beyond the immediate control of the manufacturer (e.g. particular procedures to be followed in operating the device, training and qualifications of users, etc.). More importantly, the manufacturer usually does not have detailed information about the specific environment and the processes within which the device will be operated within a particular health-care provider’s setting. In complex systems, this is a serious cause for concern, as there may be unintended interactions that may not have been accounted for.14,21
The regulatory process should provide better transparency in the decision-making process about the risks that patients are subjected to from medical devices, and there should be greater active involvement of health-care providers in the assurance of safety of their services that involve medical devices. Improved communication among manufacturers, regulators and service providers and clinicians is an essential prerequisite for this.
The two NHS safety standards Data Set Change Notice (DSCN) 14/2009 22 and DSCN 18/2009 23 are addressed to manufacturers of health IT products and service providers employing such products, respectively. Similar to IEC 80001, the aim of these standards is to introduce a risk management process over the life cycle of the health system based on ISO 14971, and to bridge the gap in communication between manufacturers and service providers.
These NHS standards additionally introduce and place emphasis on the concept of Clinical Safety Cases. This is derived from consideration of best practice in other safety-critical industries, such as defined in the UK Ministry of Defence standard Def-Stan 00-56.
24
The Clinical Safety Case is
an argument, supported by a structured body of evidence, in the clinical risk management file, that provides a compelling, comprehensible and valid case that a system for deployment and use is, as far as the clinical risk management process can realistically ascertain, free from unacceptable clinical risk for its intended use.22,23
The two standards require that both the manufacturers and the service provider develop such a Clinical Safety Case. The reports summarising the Clinical Safety Cases are an important communication tool between manufacturers, service providers and other stakeholders. The concept of safety cases as a regulatory tool utilised in many safety-critical industries and its potential application in health care are discussed in the next section.
Life-cycle safety argument to facilitate communication and to enhance transparency
Safety cases
We reviewed regulatory practices in six safety-critical industries (air traffic services, automotive, defence, nuclear, petrochemical and railways) in the United Kingdom. 16 In these safety-critical industries, manufacturers and operators of systems have to provide evidence of adequate safety performance of their systems to the respective regulatory authorities. The way this is done has changed significantly over the past 20 years, predominantly in response to major accidents and changes to the economic environment (e.g. the privatisation of railways leading to a fragmented industry and mixed economy). Previously, manufacturers and operators claimed safety through satisfaction of specific standards and technical requirements specified by the regulator. However, this has proven to be an ineffective and inefficient way of safety management. Current approaches require that manufacturers and operators demonstrate that they have adopted a thorough and systematic process to understand proactively the risks associated with their systems and to control these risks appropriately. They still need to demonstrate compliance with any applicable requirements specified by the regulator, but this approach goes beyond the reactive and standards-based approach to safety management.
In the United Kingdom, these duties are often fulfilled through the use of safety cases. The purpose of a safety case is to provide ‘a structured argument, supported by a body of evidence that provides a compelling, comprehensible and valid case that a system is acceptably safe for a given application in a given context’. 24 The core of the safety case is typically a risk-based argument and corresponding evidence to demonstrate that all risks associated with a particular system have been identified, that appropriate risk controls have been put in place and that there are appropriate processes in place to monitor the effectiveness of the risk controls and the safety performance of the system on an ongoing basis. In addition to such a risk-based argument, recent research suggests the safety case should also communicate the amount of confidence that one can have in the safety argument. 26 The use of safety cases is an accepted best practice in UK safety-critical industries, and is adopted by companies as a means of providing rigour and structure to their safety management systems.
Purely textual accounts of safety justifications often make it difficult for the reader or assessor to follow the logical argument that relates evidence to the claim it is intended to support. In addition, multiple cross-references make such documents generally hard to read and difficult to communicate to stakeholders of different backgrounds. Graphical argument notations, such as the Goal Structuring Notation (GSN) 27 and Claims–Arguments–Evidence (CAE), 28 explicitly represent the key elements of any safety argument, and their relationships. Tools that facilitate the construction of such graphical representations have been developed ((Safety Argument Manager (SAM), Adelard Safety Case Environment (ASCE)).28,29 With these tools, the construction and the communication of safety cases may be facilitated. 30 Figure 2 provides an overview of the main GSN elements. Claims or goals can be broken down into sub-claims until these can be satisfied by reference to evidence. The reasoning behind this breakdown can be made explicit through the use of the strategy element. Contextual elements serve to frame a particular claim by explicitly stating the context within which the claim is assumed to be met. Justifications provide reasons why a particular strategy is selected or why a particular claim is made.
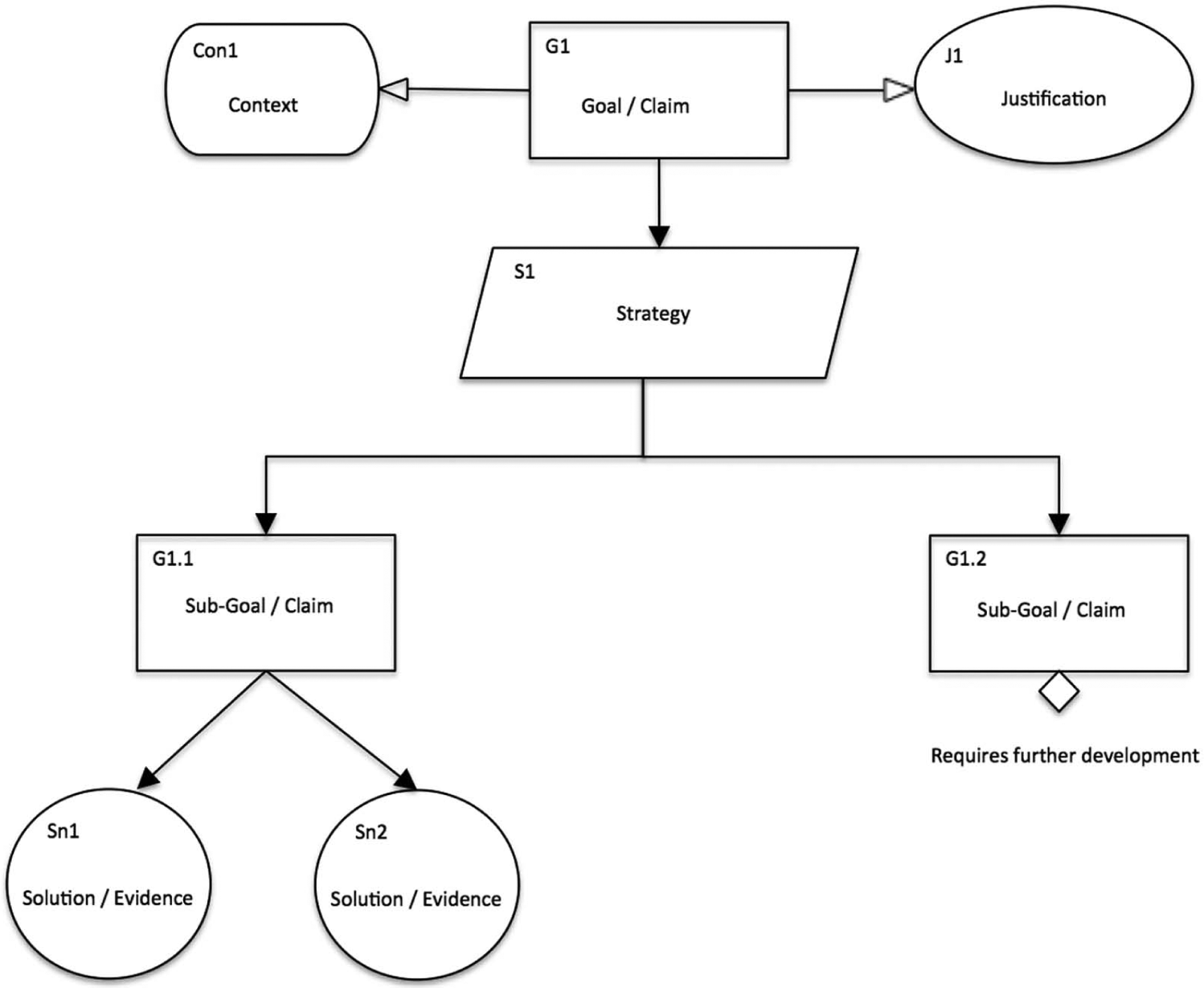
Overview of GSN elements.
Claims can be backed up with quantitative and qualitative evidence from analytical and empirical sources. Common types of evidence include, for example, descriptions of analytical hazard identification and risk assessment processes and their results, measurements and audits of system performance and relevant parameters, investigation reports of incidents and adverse events, action plans and minutes, staff surveys, competency assessments and so on.
Life-cycle safety argument
In order to illustrate the role that safety cases could play in facilitating communication between the different stakeholders, we outline a possible generic high-level safety argument. The focus is on the possible involvement of service providers as a potential way of overcoming the shortcomings described in section ‘Regulatory context and current practices’. Service providers require assurance that manufacturers of medical devices and health IT systems have applied sound risk management principles, and they need to be provided with information about the risks associated with the use of these systems, how these risks have been dealt with and what kind of residual risks remain.23,25 Service providers should then use this information in order to apply their own risk management processes to ensure that the systems are safe in use within the actual operational realities. In the example below, we consider the types of arguments and evidence that the different stakeholders should provide and be made aware of, rather than the development of detailed device-related hazard mitigation arguments. For an example of the latter, please see Weinstock and Goodenough, 31 where a detailed safety argument for a generic infusion pump is described.
The demonstration of safety of a medical device or health IT product should take consideration of the entire life cycle. This includes demonstration that the system design and production are safe, that the operation of the system in a particular organisational context is safe and that there are processes in place that monitor the safety performance of the system, which enable adequate response to any unforeseen situations or novel risks. This is illustrated in Figure 3, where these goals have been allocated to manufacturers and service providers, respectively (and jointly as in the case of Goal 2). Figure 3 represents this as one argument, but in practice, manufacturers and service providers will produce their own separate safety cases. The important issue is that these safety cases are informed by this overall structure, and that the different stakeholders have access to the information provided in the safety cases.
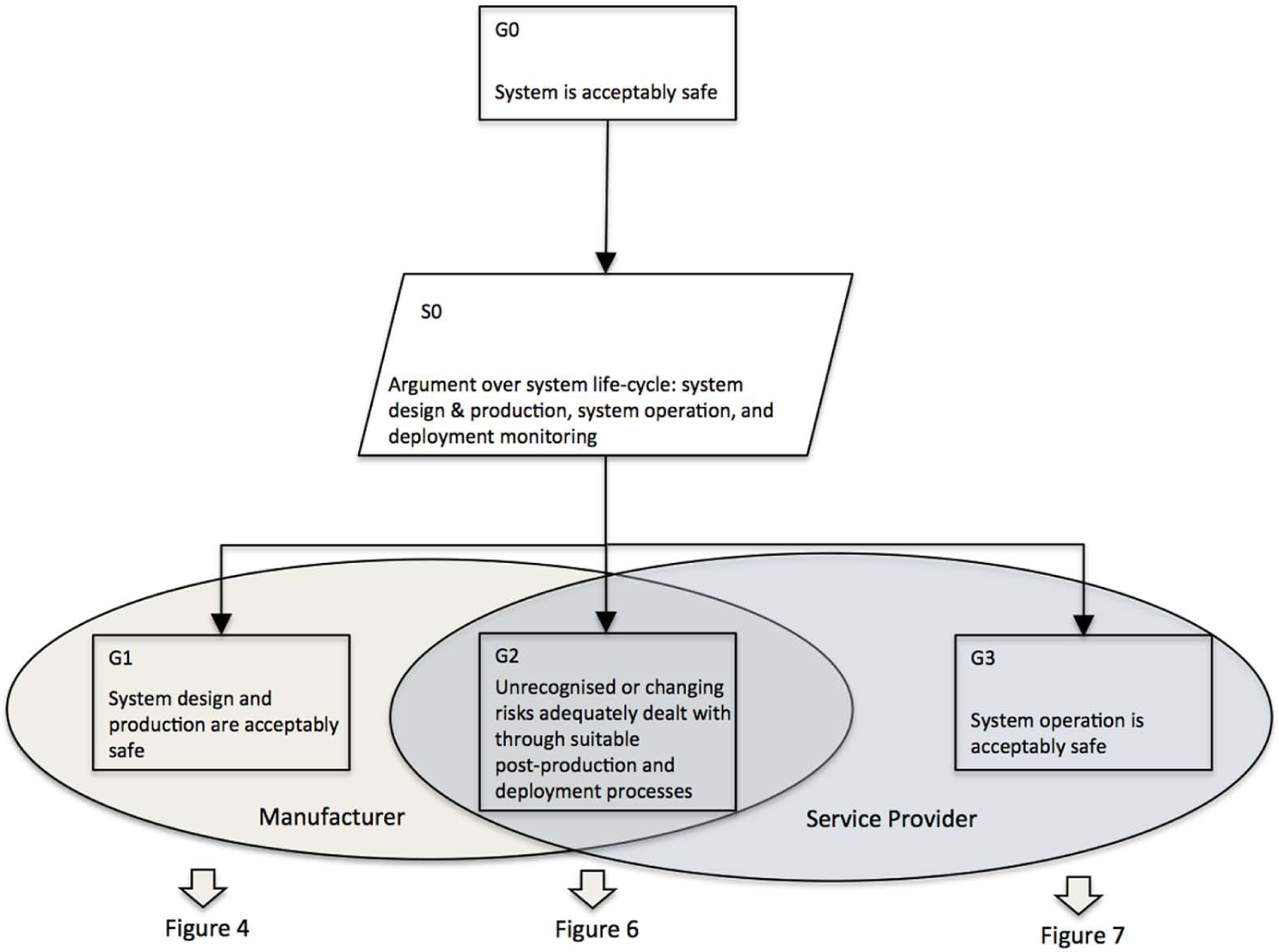
High-level safety argument over the life cycle of a medical device/health IT product.
Figures 4 and 5 provide some insights into the type of safety justification expected from manufacturers. The claim that system design and production are adequately safe is satisfied through a risk-based argument based on the principles for risk management set out by ISO 14971. This is a simplification, since manufacturers will need to comply with a range of other standards, as described in section ‘Regulatory context and current practices’. However, for the purpose of this article, this argument will suffice in order to illustrate the type of considerations that should be made in order to enable involvement of users and service providers in the overall justification of safety.
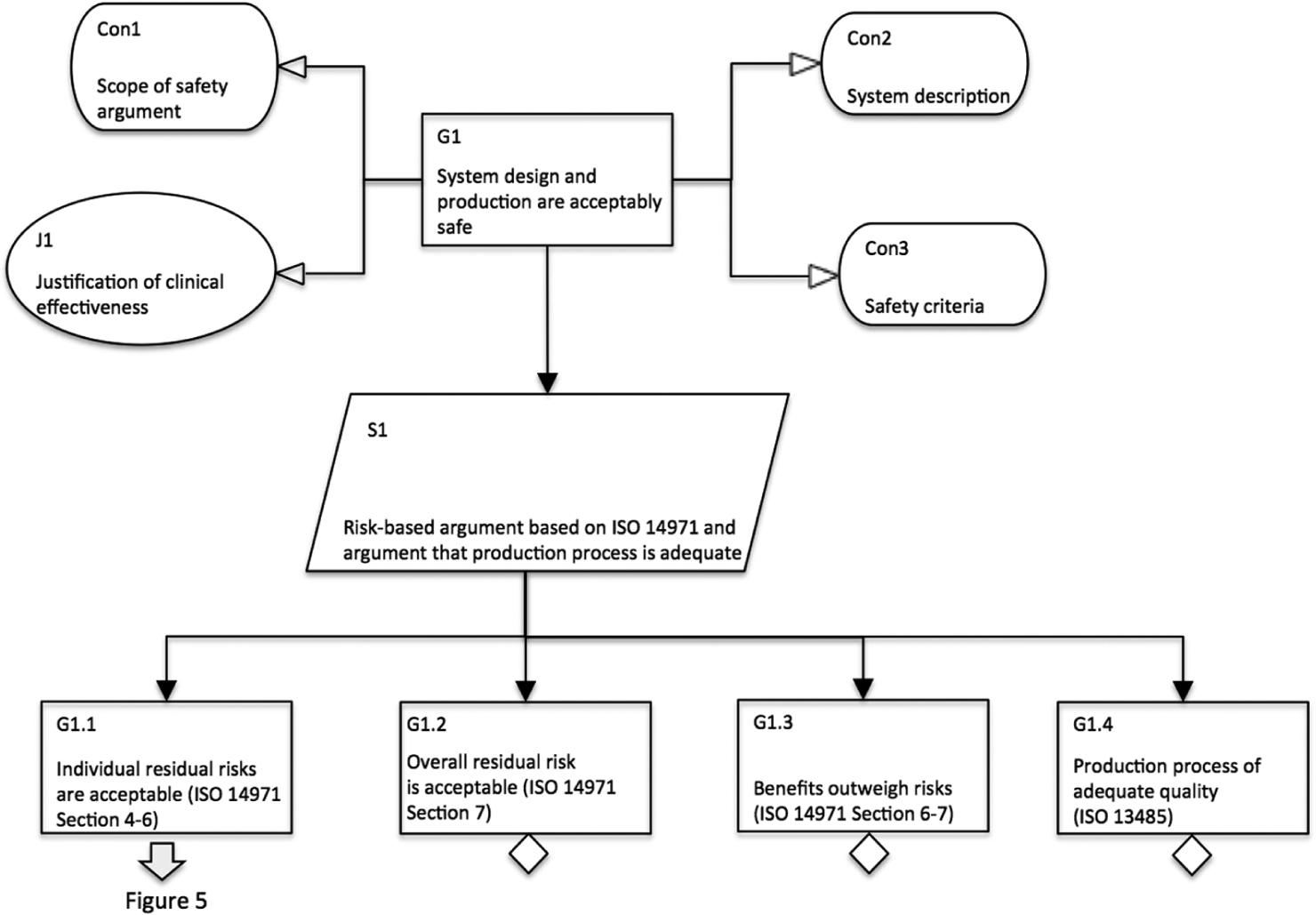
Breakdown demonstrating that system design and production are adequately safe.
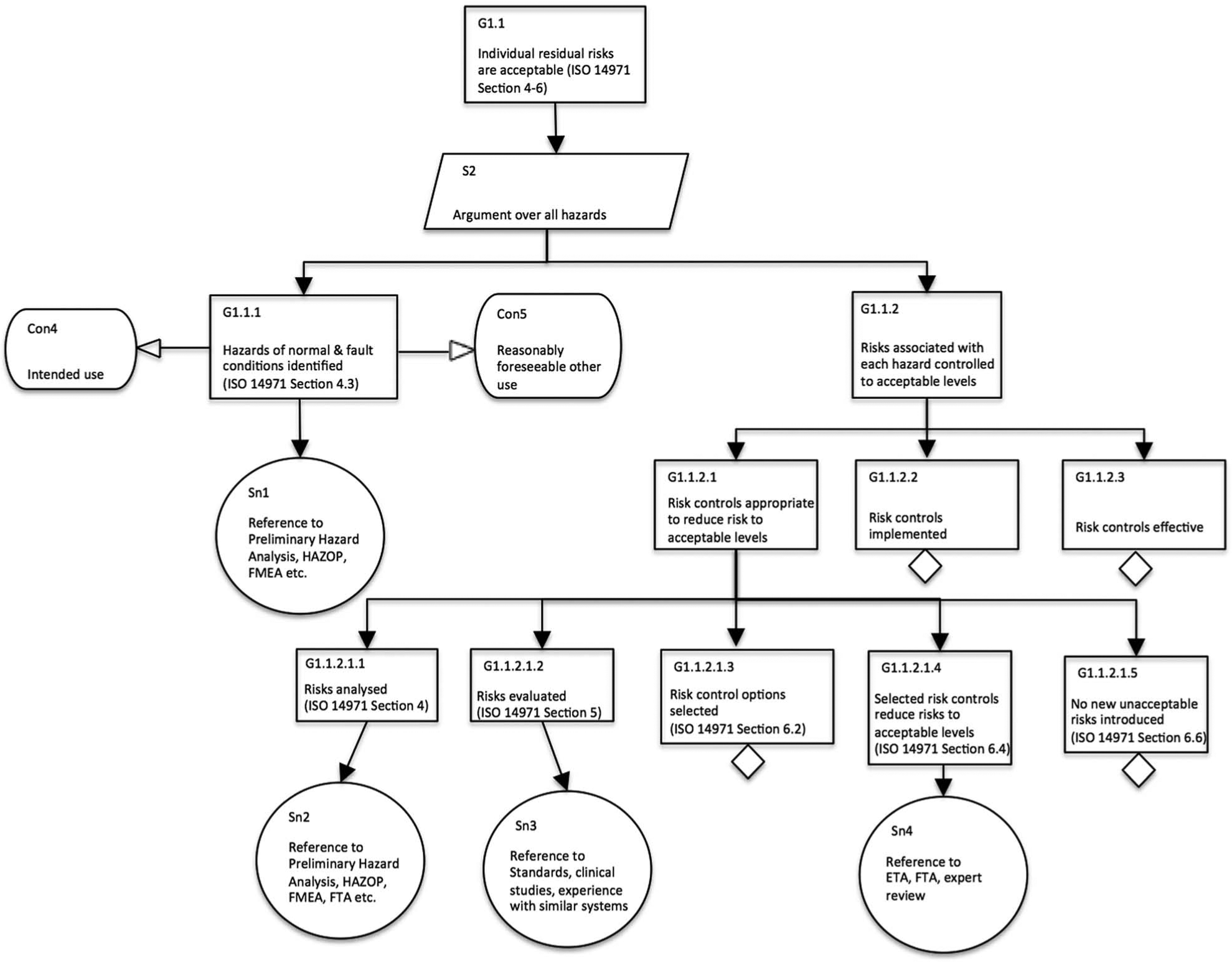
Breakdown demonstrating that individual residual risks are reduced to acceptable levels (manufacturer safety justification).
Figure 4 also contains a number of contextual elements that provide important information about the scope of the safety argument (e.g. whether a new system or a change to an existing system is considered), the system under consideration and the safety criteria that have been applied in order to determine acceptability of risk. The latter is important since the manufacturer is required to determine acceptability of risk, and setting out these criteria explicitly enhances overall transparency. Figure 4 further contains a justification of clinical effectiveness, which should be provided in order to provide a clinical reason for the system’s development.
Figure 5 provides an illustration of the further development of one of the main goals, namely, the claim that individual residual risks are acceptable. A common strategy for achieving this is to construct an argument over individual hazards and to demonstrate that the risk associated with each hazard is acceptable. Goal 1.1.1 states that all hazards of normal (i.e. fault-free) and fault conditions have been identified. The manufacturer needs to specify the intended use as well as any other ways in which the system could foreseeably be used when in operation in a particular setting. Goal 1.1.2 then goes on to demonstrate that the risk associated with each hazard is acceptable by showing that adequate risk controls have been determined (Goal 1.1.2.1), that these risk controls have been implemented either in the system or in the case of procedural risk controls in the user guidance and training specification (Goal 1.1.2.2) and that they are effective in actually reducing the risk as claimed (Goal 1.1.2.3). These activities form part of good industrial practice and will be familiar to manufacturers. The important issue we want to highlight here with this safety argument is the need to provide such an explicit argument that enables greater transparency and that allows users and service providers to create their own operational safety justification.
Figure 6 provides an outline for the claim that after deployment, any previously unrecognised or changing risks will be identified and adequately managed. This requires that manufacturers and users have appropriate communication channels in place through which alerts about risks can be distributed and received, and any other important information relating to the system can be disseminated (Goal 2.1). In addition, service providers need to have processes in place that monitor the safety performance of the system in operation, and there need to be appropriate communication channels to feed relevant information back to the manufacturer (Goal 2.2). Both manufacturers and service providers need to utilise this information to inform their own risk management processes (Goal 2.3).
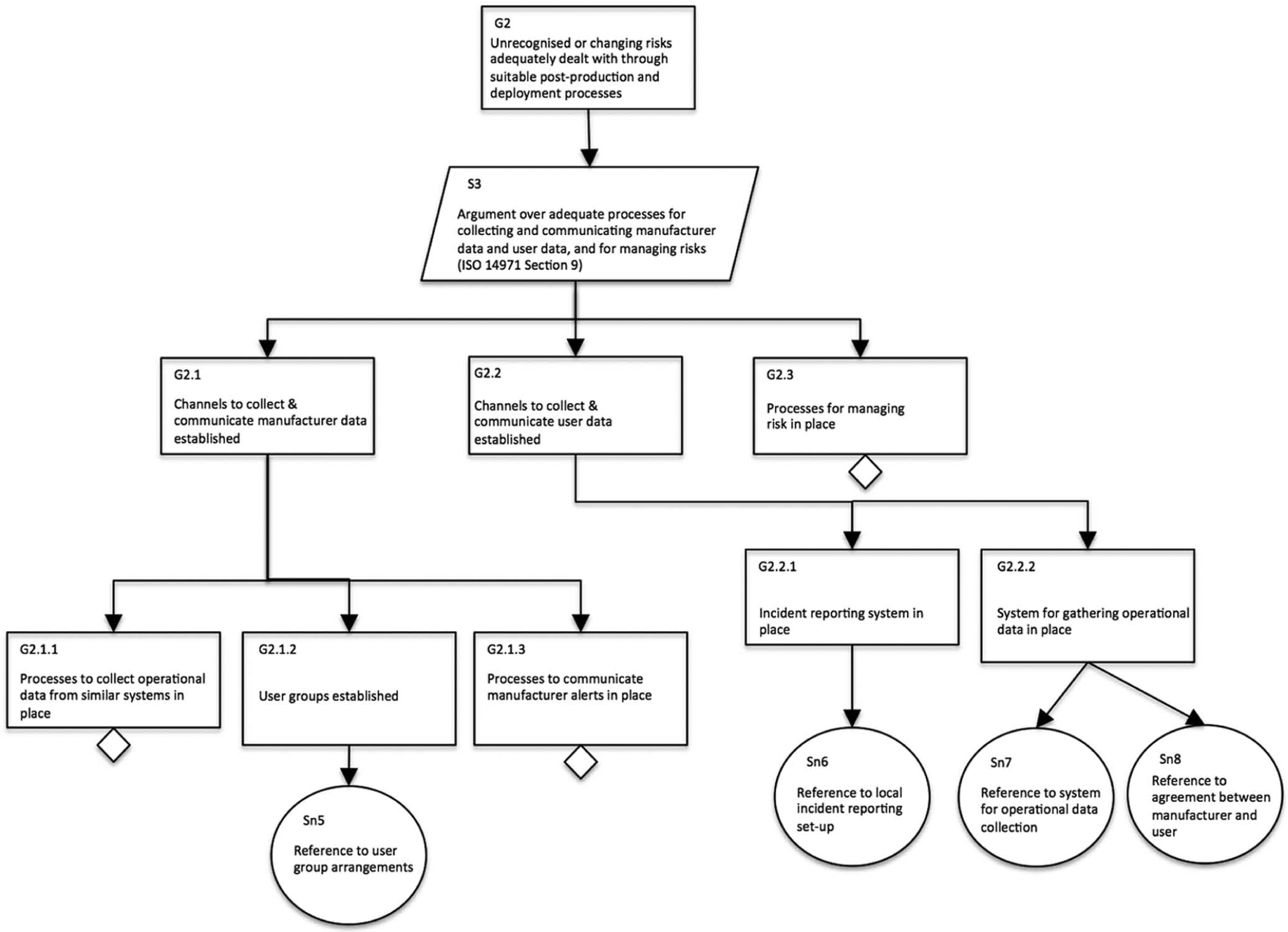
Breakdown demonstrating that previously unrecognised risks and changing risks will be dealt with adequately.
Figures 7 and 8 provide information about the operational side of the system. The service provider needs to provide assurance to themselves as well as to other stakeholders that the system in use is acceptably safe. While this safety justification follows again the principles set out by ISO 14971, there are some important considerations that are worth pointing out. In Figure 7, there are contextual elements to describe the operational context and any dependencies or interrelationships with other systems. This is an important aspect of the local risk management processes, as the manufacturer cannot have such detailed insights when constructing their own safety justification based on an assumed context. In addition, the service providers need to specify their own policies and safety criteria to determine acceptability of risk.
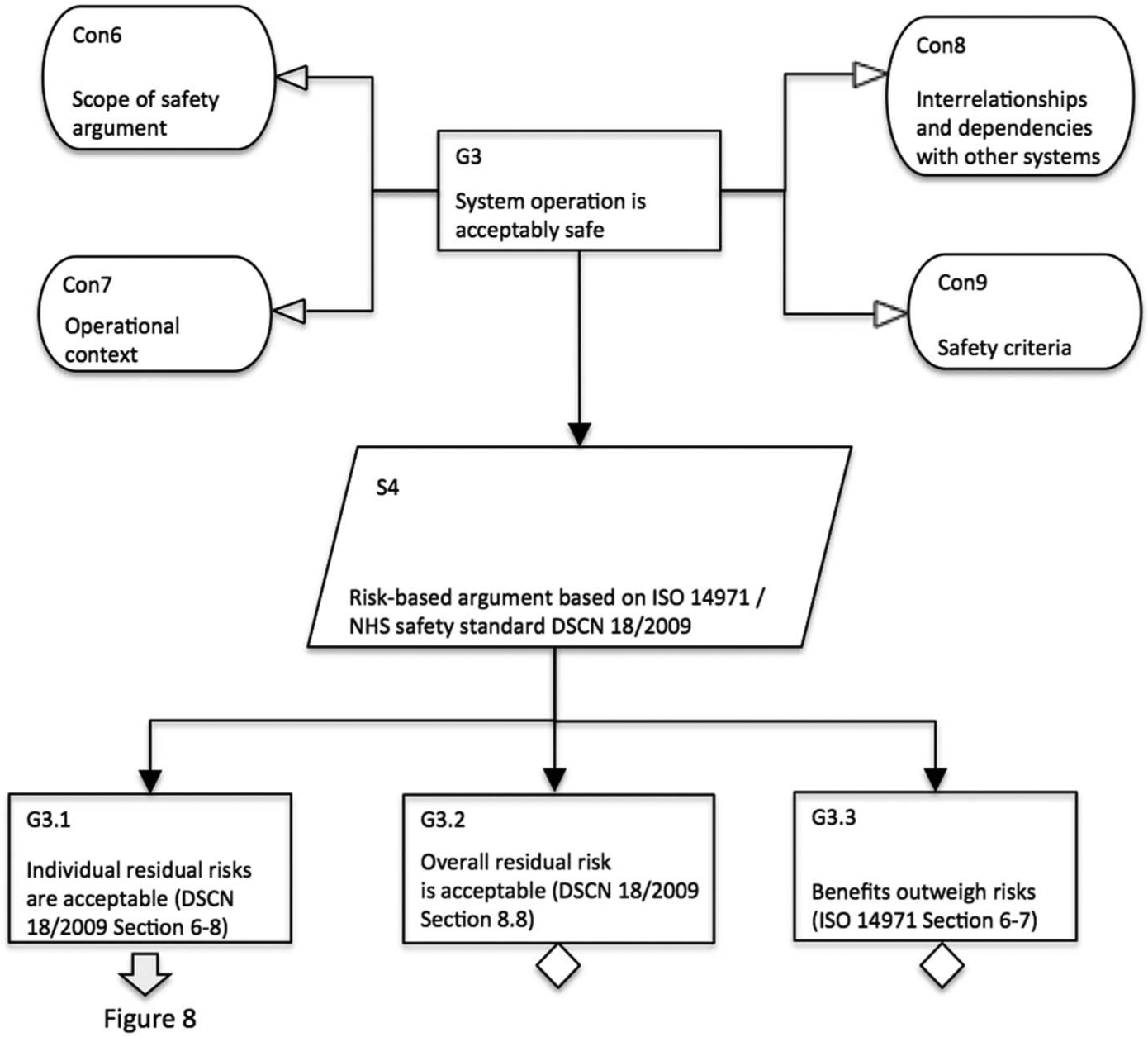
Breakdown demonstrating that system operation is adequately safe.
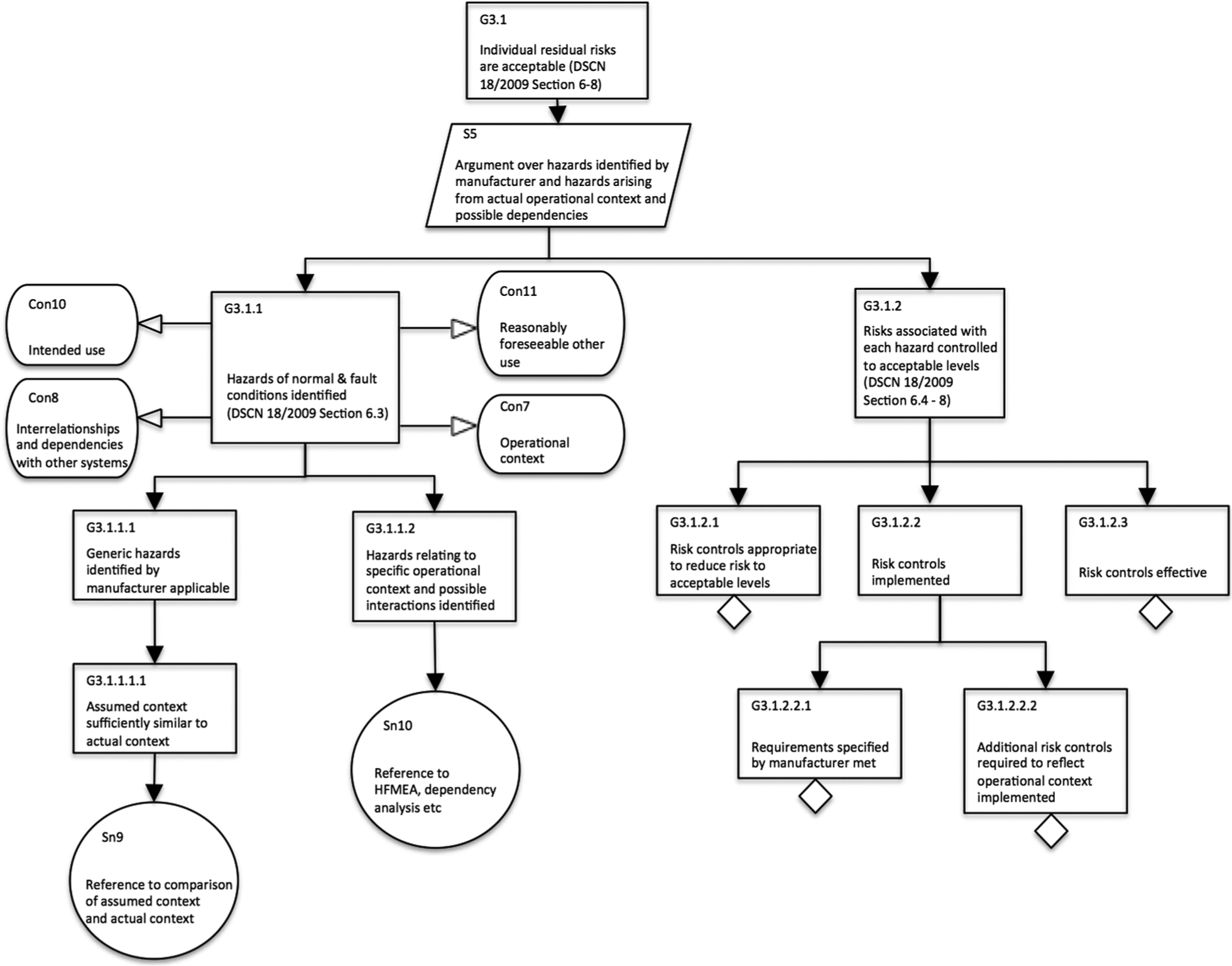
Breakdown demonstrating that individual residual risks are reduced to acceptable levels (service provider safety justification).
Figure 8 provides a breakdown for the argument that individual residual risks are acceptable. We emphasise the differences to Figure 5 (the comparable goal on the manufacturer side). In essence, the service providers need to assess the extent to which the context assumed by the manufacturer is representative of their own organisational environment. Where there are differences or additional dependencies, and interactions need to be considered, the service providers need to identify the relevant hazards through their own risk management process. In order to determine adequate risk controls for the identified hazards, service providers need to ensure that they are meeting any requirements specified by the manufacturers. These could be specific procedures or ways of operating the system, training and qualification of staff using the system, appropriate maintenance schedules and so on. In addition, service providers need to determine further risk controls as appropriate in order to ensure that the risk associated with identified hazards is reduced to acceptable levels according to their policy for determining risk acceptability.
Conclusions
The current regulatory practice in Europe is in need of change. This is likely to be a long and difficult process as the relationship between national regulators and notified bodies needs to be revised; communication and collaboration between European regulators need to be improved and the power relationships between manufacturers, regulators, notified bodies and users need to be addressed. Some people are favouring a more stringent approach as is practised for drugs and medications,2,5,13,18,32 but there are concerns that this may slow down the process of innovation and hence delay patients’ access to potentially beneficial products. 33
The focus of this article has been on lack of transparency of the regulatory process and the lack of involvement of service providers in the assurance of safety. There are some areas, such as networked medical devices, where the lack of transparency and the insufficient involvement of clinicians and service providers have been recognised, and standardisation efforts have sought to address some of these issues. Unfortunately, this new thinking is not yet commonly accepted practice, and – in the case of the NHS safety standards for health IT products discussed in this article – sometimes not widely known.
Other safety-critical industries have experienced similar problems as they underwent processes of fast technological innovation or significant market changes, for example, the privatisation of the UK railways in the 1990s. Many of these industries have adopted the safety case concept as a means to ensure that manufacturers of systems, operators and other subcontractors and stakeholders follow a systematic, structured and transparent approach to safety management that ensures adequate communication and independent scrutiny.
In the United States, the FDA has issued guidance to manufacturers of infusion pumps that recommends the use of an assurance case (an extension of the safety case) as part of the pre-market notification 510(k) submission for new devices.34 The FDA believes that the assurance case approach will eventually speed up the approval process and will achieve a more systematic, coherent and consistent way of evaluating devices. However, this guidance is still directed at manufacturers only and does not address the involvement of service providers. An extension of the use of the safety case approach to include service providers, as suggested in this article, could be a useful communication tool in the assurance of safety of medical devices and health IT products.
Challenges
The review of regulatory practices in other industries 16 has shown that there are a number of challenges that need to be overcome in order to ensure that the use of safety cases achieves its aims. Some of the challenges related to the adoption of safety cases that need to be addressed include the following:
Becoming a paper exercise. Safety cases must not become just another ‘filed return’. The production of a safety case is an opportunity for gaining greater understanding of the current picture of safety, and for potentially making safety improvements.
Being removed from everyday practice. Safety cases are supposed to address the realities of everyday system operation. It is important that they do not become a desk exercise that relates only dimly to actual practice. The primary concern of a safety case should lie in demonstrating safety, rather than being an exercise in attempting to shift liability, or in merely demonstrating compliance with ‘due practice’.
Being produced by the wrong people. Organisations may be tempted to outsource the production of safety cases to external consultants. This would defeat the purpose of a safety case of ensuring that organisations themselves consider the risks associated with their systems in a systematic and thorough way. Safety case development needs to involve all of the relevant stakeholders with an understanding of, and involvement in, what actually makes systems safe (or unsafe).
A way forward?
Safety cases could be a useful tool to facilitate communication between stakeholders in the assurance of safety of medical devices and health IT products. They document the rationale behind activities, list assumptions and limitations, and provide transparency and confidence in the safety activities and results. In industry, regulation is usually the main driver for the adoption of safety cases, and change to such processes is often slow. There is a need for continuing education in systematic and structured safety practices in particular not only on part of the service providers who will have to take greater responsibility in assuring the safety of devices and products in use but also on part of the regulator who needs to review and scrutinise safety documentation and ultimately provide confidence to the public. The benefits of the adoption of safety cases need to be demonstrated in targeted case studies and pilots. At present, there is little empirical evidence and experience about how safety cases in health should be constructed, and the types of arguments that should be made. Safety cases in health care may need to include arguments about clinical effectiveness, economics and patient throughput that are particular to the health sector. The use of safety cases as a regulatory instrument to facilitate the regulatory process and to ensure that feedback provided to organisations is clinically relevant should be investigated further building on the recent experiences of Connecting for Health in the UK NHS (health IT) and the FDA in the United States (infusion pumps).
Footnotes
Acknowledgements
Robin Bloomfield, David Embrey, Jamie Henderson and Alberto Pasquini were part of the research team. George Cleland, Ibrahim Habli and John Medhurst provided input to reviews on regulatory practices in health care, the automotive industry and railways. We acknowledge the discussions with members of the Medical Devices Group of EWICS TC7. We are grateful to the anonymous reviewers for their comments and suggestions for improvement.
Funding
This work was funded in part by a research grant from the Health Foundation (Registered Charity Number: 286967).