
Abstract
Background
RAY1216 is an alpha-ketoamide-based peptide inhibitor of severe acute respiratory syndrome coronavirus type 2 (SARS-CoV-2) major protease (Mpro). This study evaluated the absorption, distribution, metabolism and excretion of [14C]-labelled RAY1216 by oral administration.
Research design and methods
This phase Ι study was designed to assess the pharmacokinetics, mass balance and metabolic pathways in 6 healthy Chinese adult men after a single fasting oral administration of 240 mL (containing 400 mg/100 μCi) [14C] RAY1216.
Results
RAY1216 absorbed rapidly in the plasma, with a Cmax of 1796.83 ng/mL, tmax of 1.42 h and t1/2 of 5.97 h. RAY1216 is mainly excreted through feces and a small amount through urine, indicating that the excretion of RAY1216 occurs through the fecal route, within 96 h after administration, the majority (>90%) of the radioactive substances were excreted 104.17% of the metabolites were identified in urine and fecal samples. The radioactive transformation pathways suggest that RAY1216 has multiple metabolic pathways, including Oxidation-dealkylation, Mono-oxidation, Hydrolysis, and Urea binding. There were no reports of death, serious adverse events (SAEs), or withdrawals related to SAEs.
Conclusion
The overall recovery rate data of radioactive substances in the excreta of all 6 subjects indicate that favourable mass balance recovery. The overall safety profile is favourable, and it demonstrates promising potential in mitigating both the duration and severity of COVID-19, and the comprehensive clinical safety and therapeutic effect are significantly superior to those of similar COVID-19 treatment drugs. RAY1216 can be referred to and further verified for the Phase II and Phase III clinical trials for the treatment of COVID-19.
Introduction
COVID-19 is caused by severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2). Variants such as SARS-CoV-2 Omicron, which has highly mutated spike proteins, have rapidly emerged as a result of population immune pressure established by infection and vaccination.1–3 Small molecule antiviral medications are the main therapeutic drugs for SARS-CoV-2 because of their easy absorption, simple penetration of cell membranes, and high safety.4,5
Remdesiviv, which was the first drug approved by the Food and Drug Administration to combat COVID-19, inhibits RNA transcription by binding mainly to viral RNA-dependent RNA polymerase (RdRp).6–8 However, as clinical trial data accumulated, this drug was found to have no significant effect on hospitalized patients with COVID-19.7,9 Therefore, in 2022, the WHO recommended that Remdesiviv should be used only for patients with light-to-moderate COVID-19. 10 Similarly, Nematrivir/Ritonavir has an adverse drug interaction profile (i.e. inhibition of CYP3A4, CYP2D6, P-gp, and OAT1B1) may give rise to various security risks. 11 Nirmatrelvir also need to be used in combination with Ritonavir. There is a risk of drug interactions both during the infection period and after discontinuation. Therefore, during the infection period, it is prohibited to use it in combination with some anti-infective drugs, bronchodilators, etc., to avoid affecting the clinical treatment effect of some patients with complications (such as asthma and cancer).12,13 After discontinuation, due to Ritonavir’s inhibition of the activity of enzymes such as CYP3A4, the potential risk rate of drug interactions in patients after discontinuation of Nirmatrelvir/Ritonavir is higher than 50.0%. 12 However, the in vitro pharmacodynamic results of RAY1216 indicated that RAY1216 had a remarkable inhibitory effect on Mpro and had significant antiviral activity against different SARS-CoV-2 variants (wild-type, Alpha, Beta, Delta, and Omicron). RAY1216 can be administered as a monotherapy, exhibiting a relatively low risk of off-target effects, favourable pharmacokinetic properties and a high safety profile. Phase I clinical trials demonstrated that oral absorption of RAY1216 tablets was rapid, and the exposure increased with the dose. After multiple administrations, the drug reached a steady state quickly, showing good human pharmacokinetic characteristics and safety. 14 Therefore, we hypothesize that the rapid absorption of RAY1216 observed in humans may be associated with its primary excretion route via feces. Phase III clinical trials showed that RAY216 could significantly shorten the time to sustained clinical recovery of 11 target symptoms in adult patients with mild to moderate COVID-19 infection, and rapidly and significantly reduce the viral load of the novel coronavirus compared with the placebo group. 15 Therefore, we could investigate whether RAY1216 is an effective oral treatment for COVID-19.
Radioactive-labelled absorption-distribution-metabolism-excretion (ADME) studies provide the most comprehensive data set for understanding how the human body processes drugs and are considered the gold standard for small molecule drug ADME because they can quantitatively determine the parent drug, metabolites and total drug-related substances simply and accurately independent of the compound structure.16–19 Therefore, this method might be widely applicable. However, less mass balance exists to assess COVID-19 therapeutics. 11 RAY1216 shows better efficacy in certain specific populations (such as those with compromised immune function), but its efficacy may vary in other populations, and the data on its long-term safety and effectiveness are still limited. 15 It is necessary to investigate the metabolic pathways and safety data of [14C]-labelled RAY1216. Furthermore, in this experiment, 100 μCi of [14C] was used, which met the ICH/FDA requirement of ≥90% recovery. 16 The radiation dose was only 0.021 mSv (ICRP limit of 2%), so the use of radioactive labelling was also reasonable. 18 This trial thus partially addresses some of the gaps in the mass balance of COVID-19 treatments.
This study aims to conduct a more comprehensive Phase Ι clinical trial on 6 healthy subjects to assess the mass balance of RAY1216. The main research objective is to evaluate the substance balance after a single fasting oral administration of 240 mL of a drug containing 400 mg/100 μCi [14C]RAY1216, and to determine the main metabolites, pharmacokinetics, and safety.
Method
Research materials
RAY1216 (batch number, 230101; purity, 98.7%) and the drugs used were provided by the Guangdong Xianqiang Pharmaceutical Co., Ltd [14C] RAY1216 (chemical purity, 99.6%; batch number, NH00034-022-20230710; specific activity, 47.06 mCi/mmol; radiochemical purity, 99.7%) was provided by the Shanghai Pharmatech New Drug Development Co., Ltd [14C] RAY1216 was prepared as a sealed powder preparation in glass bottles, each containing a total amount of 400 mg/100 μCi, prepared with ∼240 mL of normal saline before use. [14C] RAY1216 was prepared and packaged by the Nanjing Pharmaceutical New Drug Development Co., Ltd, China. The basic structural formula of [14C] RAY1216 is shown in Figure 1. Basic structural formula of [14C] RAY1216.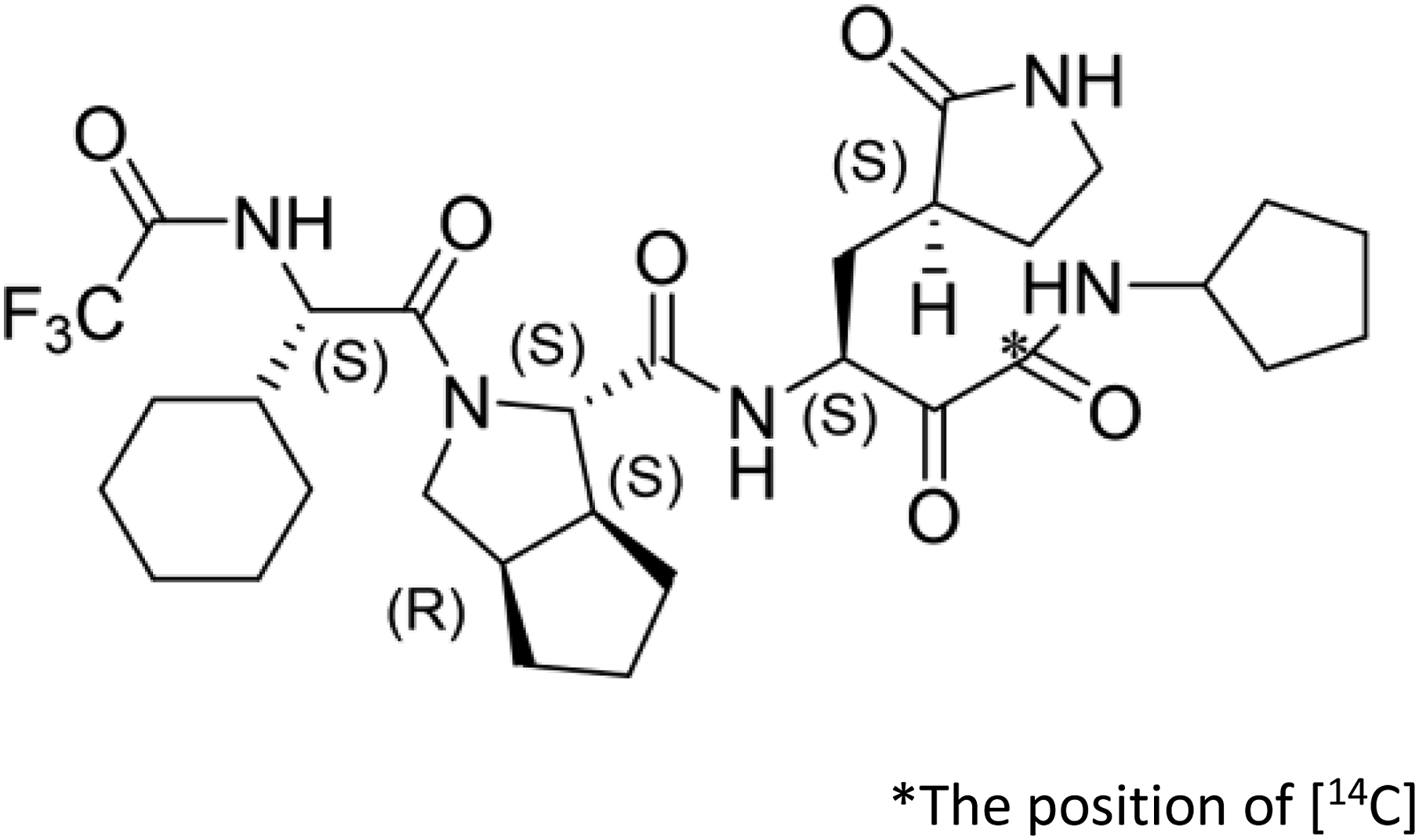
Justification of dose
This clinical trial evaluated the mass balance, metabolic characteristics and metabolite identification of [14C] RAY1216, as well as the safety profile of an oral dose of 240 mL containing 400 mg/100 μCi of [14C] RAY1216. Preclinical data show that a single dose of RAY1216 can achieve an effective exposure level, and the No Observed Adverse Effect Level (NOAEL) for animals is significantly higher than the equivalent dose for humans. This dosage is similar to that of the similar drug Obeldesivir, which uses a [14C]-labelled dosage of 500 mg/100 μCi. 11 According to the tissue distribution and substance balance data of [14C] RAY1216 in rats, the tissue‒organ equivalent dose and the whole-body effective dose of [14C] RAY1216 in healthy adult male subjects were determined using the tissue-weighting parameters specified in ICRP 103 20 to ascertain the actual [14C] RAY1216 equivalent received by each subject. The final estimate of [14C] RAY1216 in a single oral administration of 100 μCi (∼3.7 MBq) for adult physically healthy men showed that, after a single oral administration of 240 mL [14C] RAY1216 with 400 mg/100 μCi, the total body effective radiation dose was approximately 2.12 mrem (0.0212 mSv). This is approximately 0.071% of the 3000 mrem exposure limit set by the FDA 21 and 2.12% of the 1 mSv limit allowed by the International Commission on Radiological Protection (ICRP) 22 (risk class IIa, 1/5 of the annual patient dose limit set by the Swiss Radiation Protection Regulations).
Clinical study design
This trial adopted a single-center, single-dose and open-label trial design, randomization or blinding was not implemented (Figure 2). The questionnaire and methodology for this study (trial registration: NCT06362460) was approved by the Human Research Ethics committee of the University of Bengbu Medical University First Affiliated Hospital (Ethics approval number: 2023148). The study was performed in accordance with the ethical standards as laid down in the Declaration of Helsinki and its later amendments or comparable ethical standards. All protocols, amendments and consent forms were reviewed by the Institutional Review Board (IRB) or Ethics Committee (EC).
The trial was designed to enrol six to eight healthy men who would receive a single oral administration of 240 mL (containing 400 mg/100 μCi) of the study drug [14C] RAY1216 on the morning of day 1 under fasting conditions. The subjects were required to take the drug after an overnight fasting period of at least 10 h, with no water intake for 1 h prior to and 1 h following drug intake and fasting for 4 h after drug intake, a standardized low-fat meal (50–60% carbohydrates, 20–25% protein, and 20–25% fat) was provided to all subjects 4 h following medication administration.
Blood, urine, and fecal samples for the analysis of [14C] RAY1216 in plasma; the overall radioactivity in whole blood, plasma, urine, and feces; and metabolite examination and structural identification in plasma, urine, and feces were collected before and at specified time points within 336 h after the dose was administered at the study center. Mass balance analysis of [14C] RAY1216 was also conducted. After the administration of the drug is completed, the research unit will return the used empty vials and the remaining unused investigational drug to Nanjing Wuxi AppTec New Drug Development Co., Ltd for the determination of residual radioactivity. After that, Nanjing Wuxi AppTec New Drug Development Co., Ltd will be responsible for the unified destruction and issue a destruction certificate.
All biological samples were systematically collected at the preset time points. Special containers were used, and the samples were separately frozen and stored, as well as subjected to incineration treatment. The entire process was carried out under refrigerated conditions and the temperature was recorded to ensure no radioactive cross-contamination.
If the total radioactive recovery rate collected over two consecutive days is higher than 90%, or the radioactive content in urine and feces is lower than 1%, or the plasma radioactive concentration at two consecutive sampling points is lower than three times the background plasma value, the researcher will decide to stop sample collection for the subject based on the radioactive detection results. If the subject vomits after administration, all vomitus will be collected and tested for total radioactivity throughout the study period.
Sample collection and processing
Blood samples were collected within 30 min before administration and within 168 h after administration in EDTA-K2 anticoagulated collection tubes. The plasma was obtained by centrifugation for analysis of whole blood and total plasma radioactivity, identification of plasma metabolites, and pharmacokinetic analysis of RAY1216 and its metabolites. All EDTA-K2 anticoagulated collection tubes were stored in a refrigerator at approximately -80°C until delivery to the bioassay and Radioassay Supplier (Wuxi Apptec New Drug Development Co., Ltd, Nanjing, China).
Quantitative collection of excreted urine samples was conducted within the time interval of 24 h before administration and 336 h after administration for the analysis of RAY1216, determination of total radioactivity, metabolite profiling and structural identification. According to the ratio of urine volume: 20 mg/mL (polyethylene glycol octyl phenyl ether) Triton X-100 solution volume = 99:1, the prepared 20 mg/mL Triton X-100 solution was immediately added, mixed well, placed at 25°C for 10 min, and shaken 2–3 times. The pooled urine was transferred to a large urine bucket and stored in a 2 °C–8°C refrigerator prior to transport. Fecal samples were quantitatively collected at time intervals within 24 h before administration and 336 h after administration for the analysis of RAY1216 in feces, determination of total radioactivity, and metabolite analysis and structural identification. The collected feces were placed in labelled boxes and stored at −80°C in a refrigerator before transportation.
If vomiting occurred after dosing, the vomit was collected throughout the study. After each analysed, the vomit was placed in a labelled collection box and stored in a refrigerator at −80°C (−60°C to −90°C).
Research population
The inclusion criteria for healthy adult male subjects were as follows: aged 18–45 years, weighing at least 50 kg, and having a body mass index (BMI) between 18 and 28 kg/m2. The screening criteria were those who worked in conditions requiring long-term exposure to radiation or who had notable radiation exposure (≥2 chest/abdominal CT or ≥3 other X-ray examinations) within 1 year before the trial. Habitual constipation or diarrhoea, defecation less than once a day, hemorrhoids or perianal disease, and gastrointestinal dysfunction, such as irritable bowel syndrome, may affect the use of medication. Subjects with difficulty swallowing the study drug or a history of Gastrointestinal disorders that severely affected drug absorption were also excluded.
The subjects had not taken any combination of prescription drugs during the 14 days before screening, including vitamin products, health medicines and Chinese herbal medicines. Strong CYP3A4 inhibitors, inducers, or substrate drugs sensitive to CYP3A4 or CYP2C19 and P-gp inhibitors or inducers must not be used from 28 days before screening and throughout the study. Subjects with any disease or condition, including gastrointestinal, renal, hepatic, neurological, hematological, endocrine, oncological, pulmonary, immunological, psychiatric, cardiovascular, or cerebrovascular disorders, which the investigators considered likely to impact the trial results, were also excluded.
Sample measurement
Measurement of RAY1216 and its metabolite concentrations in plasma
Plasma concentrations of RAY1216 were determined at WuXi AppTec (NJ, JS, China) using a validated liquid chromatography-tandem mass spectrometry (LC-MS/MS) bioanalytical method. The lower limit of quantification (LLOQ) for both analytes was 5.00 ng/mL. Verapamil hydrochloride (MedChemExpress, Edison, NJ, USA) was used as the internal standard for RAY1216. All plasma samples were vortexed to mix evenly. Then, 50 μL was taken and 50 μL of (acetonitrile/water) (1:1, v:v) was added. The mixture was shaken at 1200 rpm for 1 min, and 300 μL of (methanol/acetonitrile) (1:1, v:v) was added and shaken at 1200 rpm for 5 min. The mixture was then centrifuged at 4000 rpm and 12°C for approximately 10 min 50 μL of the supernatant was taken and 200 μL of (methanol/water) (1:1, v:v) was added. The mixture was shaken at 1200 rpm for 1 min and then ready for analysis. Three batches of analysis were conducted to examine the quality control samples of RAY1216 at low, medium and high concentrations, with six samples for each concentration. If there are values below the LLOQ in each concentration sample, they will be regarded as 0, not included in the calculation of the average value and standard deviation.
RAY1216 and its internal standard were isolated from plasma samples by protein precipitation and separated on a reversed-phase chromatographic column. Quantitative analysis of the analytes was performed using an electrospray ionization (ESI) mode on a triple quadrupole mass spectrometer. The liquid chromatography system was a Waters Acquity UPLC (Waters, Milford, MA, USA) with a BEH C18, 1.7 μm × 50 mm column, injection volume: 10 μL, gradient elution (mobile phase A: water with 0.05% ammonia water); mobile phase B: methanol/isopropanol (4:1, v/v), flow rate: 0.500 mL/min,column temperature: 50°C. The mass spectrometer was an AB-Sciex triple quadrupole API 5000 with electrospray ionization.
For each batch, there are 3 concentration levels: low (15 ng/mL), medium (2000 ng/mL), and high (3750 ng/mL). Each concentration has 6 replicates. A total of 18 QC samples are prepared for each individual analysis batch. For each biological sample (plasma, urine, feces), 2 parallel extractions are performed and run twice on the instrument for liquid chromatography coupled with radioactivity detector and high-resolution mass spectrometry (LC-RAM/HRMS) identification. The biological repetition is 6.
Measurement of total radioactivity
The total radioactivity analysis in plasma, blood, feces and urine was conducted by WuXi AppTec (NJ, JS, China). The plasma samples were thoroughly vortexed. Two parallel samples (about 0.3 g each) were weighed and placed in 7 mL scintillation vials, and 5 mL of scintillation fluid was added to each vial and mixed well. The urine samples were shaken or stirred to mix well. Two parallel samples (about 5 g each) were weighed and placed in 20 mL scintillation vials, and 15 mL of scintillation fluid was added to each vial. After they were directly measured using a liquid scintillation counter (Perkin Elmer Instruments, Inc., Downers Grove, IL, USA). After thorough vortexing of the whole blood sample, two parallel portions (approximately 0.3 g each) were weighed and placed in combustion boats. After full combustion of whole blood samples in an oxidation combustion instrument, the [14C] CO2 generated from the combustion was captured by scintillation liquid and then measured using a TriCarb 3110TR liquid scintillation counter. For fecal samples, an appropriate amount of 50% isopropanol aqueous solution was added for soaking and homogenization treatment, homogenization was carried out using a tissue homogenizer. After processing, two parallel samples were weighed from each sample (about 0.1 to 0.3 g each). Then, the samples were fully combusted in an OX-501 oxidation combustion instrument (R.J. Harvey Instrument Company, Tappan, NY, USA), and the [14C] CO2 generated from the combustion was captured by Permafluor®E+ and measured using a TriCarb 5110 TR liquid scintillation counter.
Before burning the samples, a combustion recovery rate test was conducted using a standard solution containing [14C] with a known radioactive concentration to verify the combustion efficiency of the oxidation combustion instrument. By comparing the radioactive concentration of the standard sample after complete combustion with that directly added to the scintillation liquid and measured by the liquid scintillation counter, the oxidation combustion recovery rate was obtained. The recovery rate test was carried out before, between (more than 20 samples), and at the end of each day’s biological sample combustion. The results of each recovery rate test were averaged to obtain the total average recovery rate for the day of sample determination. The total average recovery rate should be within the range of 90% to 105%. If the total average recovery rate is between 90% and 98%, the average recovery rate is used as a correction factor, and the radioactive concentration of all biological samples determined on that day needs to be divided by the correction factor to obtain the actual radioactive concentration of the corrected samples.
The liquid scintillation counter determination of the biological samples in the experiment was conducted using the external standard method for quenching correction. The counting time for each whole blood and plasma sample was 20 min, and for each urine and fecal homogenate sample, it was 5 min. The scintillation counter was set to automatically convert the counts per minute (CPM) to disintegrations per minute (DPM). The value of the blank sample was used as the background to correct the measured values of the biological samples in the experimental group. The background of the instrument was set to zero.
The total radioactivity of the administered preparation in each bottle was measured before administration. The residual radioactivity in the vials was subtracted to obtain the actual total radioactivity administered to each subject. Then, the actual total radioactivity administered was divided by the radioactivity specific activity of the administered preparation to obtain the actual administered dose of the test substance for each subject.
Radioactive metabolite profiling and identification of metabolites
The plasma, feces, and urine of six volunteers were mixed, respectively, by the area under the plasma concentration-time curve (AUC), the same percentage by weight and the same percentage by volume methods. Six plasma samples, seven urine and fecal samples. The plasma and feces samples were extracted three times with 3-fold (3 × v/v), 2-fold (2 × v/v) and 200 µL of acetonitrile: methanol: water (v/v/v, 3:6:1), and centrifuged for 10 min for analysis. The extraction recovery rate of the mixed plasma samples was approximately 94%, and that of the mixed feces samples was >87%. The urine samples were extracted twice with 10 mL and 400 µL of acetonitrile: methanol: water (v/v/v, 3:6:1), and the extraction recovery rate of the mixed urine samples was nearly 90%.
The structures of the main metabolites in plasma, urine and fecal samples were analysed by LC-RAM/HRMS. By comparing the retention time of radioactive peaks, combining with conventional metabolic pathways, the ratio of isotope peaks, and mass defect filtering, the accurate mass spectrometric molecular ion peaks related to the compounds were sought, and then the possible elemental composition of the metabolites was determined and the possible metabolites were speculated. Then, the accurate molecular ions of the metabolites were subjected to secondary mass spectrometry fragmentation, and the retention time and characteristic ion fragments of the reference standard of the prototype drug were compared to determine the type of metabolites and conduct structural analysis of the metabolites.
Pharmacokinetic and statistical analysis
The primary pharmacokinetic (PK) parameter plasma analysis included the maximum (peak) drug concentration (Cmax), the time at which the maximum (peak) drug concentration was observed (tmax), Area under the concentration–time curve (AUC0-t) from first administration to last detection concentration, area under the concentration–time curve from first administration to infinity (AUC0-∞), apparent elimination half-life (t1/2), Percentage of the area under the concentration-time curve extrapolated from 0 h to the last detectable concentration (% AUCextrap), the apparent systemic clearance (CL/F) following non-intravenous administration, the bioavailability-corrected apparent volume of distribution (Vd/F); the terminal elimination rate constant (λz), and the mean residence time from time 0 to the last measurable concentration (MRT0-last) and extrapolated to infinity (MRT0-∞).
The parameters of pharmacokinetic evaluation mainly included urine and fecal excretion recovery rate and total excretion recovery rate, plasma and whole blood total radioactivity concentration ratio, the proportion of metabolite radioactivity content in urine and fecal to the total dose of administration, the proportion of circulating metabolite in plasma to the total exposure AUC (% AUC), the original drug and the total radioactive metabolite product in plasma.
All concentration data below the lower limit of quantification are presented as ‘BLQ’ in the data list and are counted as 0 in the descriptive statistics; concentration data for missing blood samples are presented as ‘NA’ in the data list and are treated as missing values in the descriptive statistics. If the %AUCextrap of the subjects is greater than 20%, then no descriptive statistical analysis will be conducted for AUC0-∞, t1/2, %AUCextrap, CL/F, Vd/F, λz, and MRT0-∞.
All PK parameters were determined via Phoenix WinNonlin (Version 8.3) software, and SAS 9.4 software (SAS Institute Inc., Cary, NC, USA) was used to statistically analyse the data.
Safety
We closely monitored subjects for adverse events throughout the study. We assessed adverse events, clinical laboratory tests (complete blood count, coagulation function, blood biochemistry, urinalysis, and fecal occult blood), vital signs, 12-lead electrocardiogram, fundus photographs, and physical examination at multiple time points from screening to study completion. Safety was analysed and assessed according to the incidence and severity of SAEs. SAEs were assessed according to CTCAE 5.0. The SAEs were described using Preferred Terms (PTs) according to the System Organ Class (SOC) classification. If the severity of an adverse event was not specified in the guidelines, the investigators assessed and tabulated the relevance and importance of the trial according to the general definition of grades 1 to 5 combined with medical judgment. Flowchart of subject distribution.
Results
Demographic characteristics
Demographic and baseline characteristics of the subjects (N = 6) a (SS).
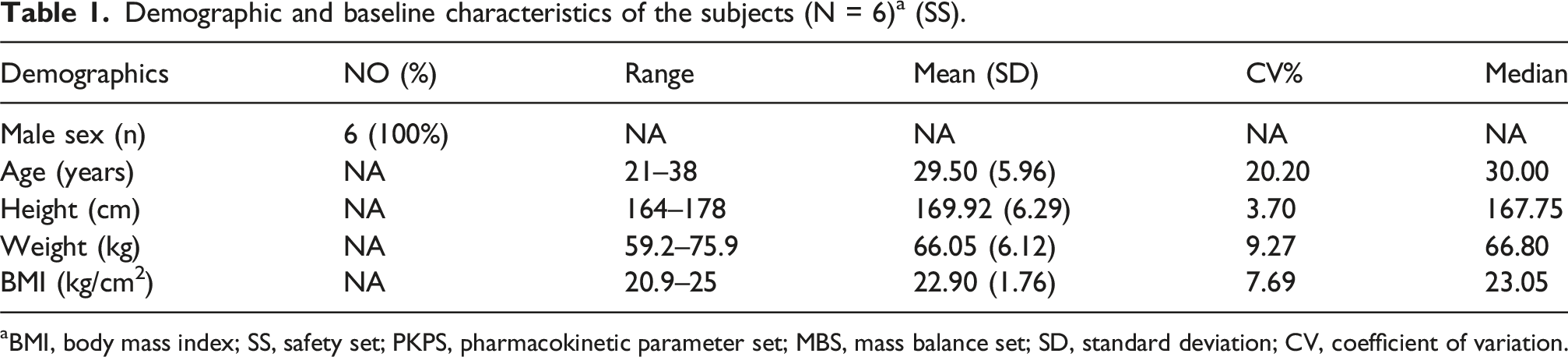
aBMI, body mass index; SS, safety set; PKPS, pharmacokinetic parameter set; MBS, mass balance set; SD, standard deviation; CV, coefficient of variation.
Pharmacokinetic
The geometric mean linear scale plasma concentration‒time curves for RAY1216 and total radioactivity are illustrated in Figure 5. After a single fasting oral dose of 240 mL (containing 400 mg/100 μCi) of [14C] RAY1216 in male subjects, it was rapidly absorbed, with both the total plasma radioactivity and RAY1216 original drug concentrations peaking within 1.5 h after administration, followed by a biphasic decrease with detectable levels within 96 h after administration.
Pharmacokinetic parameters for RAY1216 in plasma and plasma RAY1216 radioactivity; ratio of whole blood/plasma at each time point; PKPS.

aFor a subject with %AUCextrap > 20%, the parameters such as AUC0-∞, t1/2, % AUCextrap, CL/F, Vd/F, λz, and MRT0-∞ will not be included in the descriptive statistical analysis.
bThe plasma TRA concentration of four subjects was below the limit of quantification (BLQ) 24 h after administration; the whole blood radioactivity concentration and plasma radioactivity concentration of all enrolled subjects were BLQ 48 h after administration.
Abbreviations: Cmax, maximum (peak) drug concentration; AUC0-t, the area under the plasma concentration‒time curve from the first dose to the last detectable concentration; AUC0-∞, the area under the plasma concentration‒time curve extrapolated to infinity from the first dose; t1/2, apparent terminal elimination half-life; % AUCextrap,The percentage of the area under the concentration-time curve from 0 h to the last measurable concentration; tmax, the time to achieve the maximum (peak) drug concentration; MRT0-last, average residence time from time 0 to the last measurable concentration; MRT0-∞, The average residence time is extrapolated to infinity; Vd/F, bioavailability correction distribution apparent volume; λz, terminal elimination rate constant; CL/F, significant systemic clearance after non-intravenous injection.
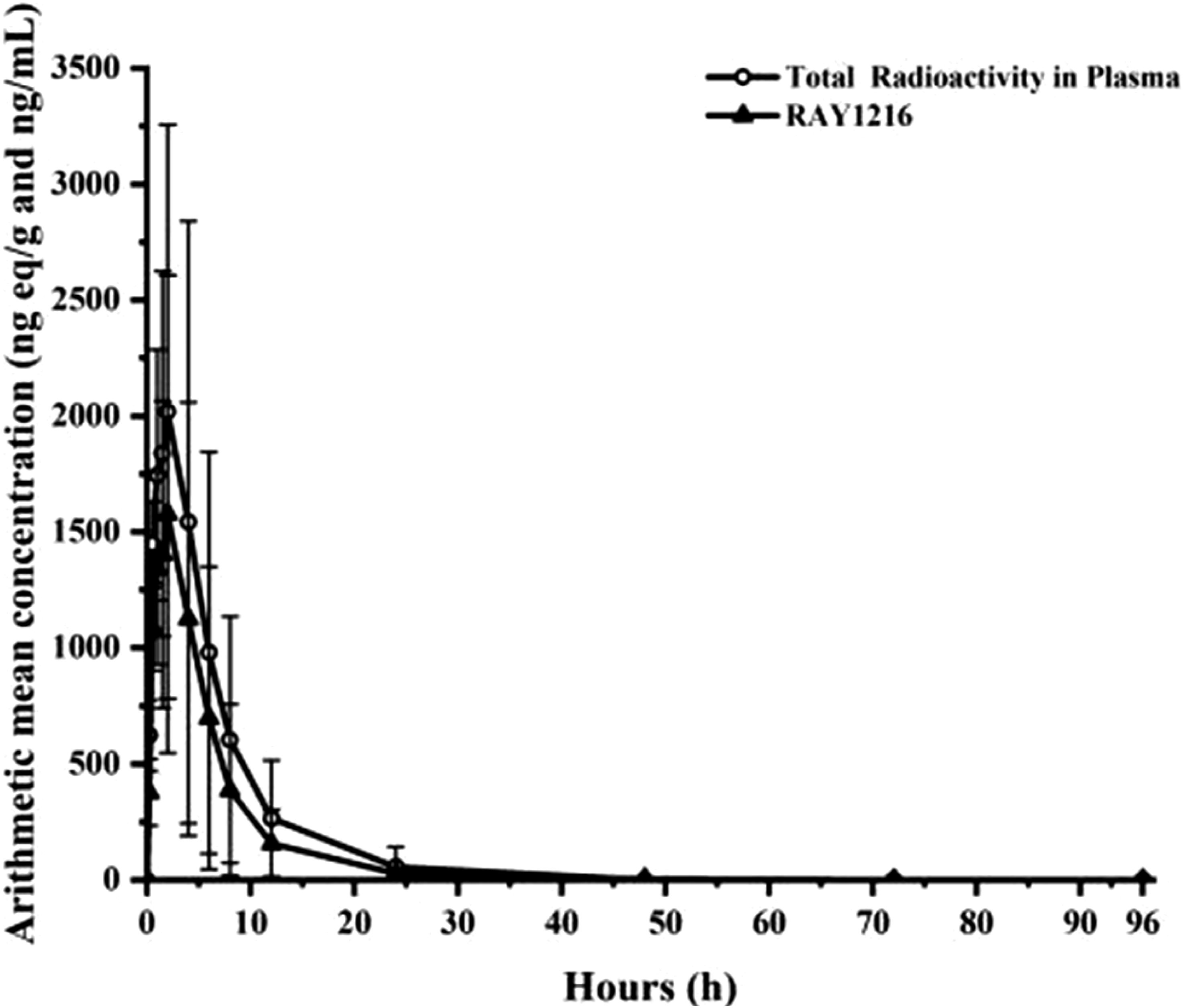
Mean (±SE) plasma concentration‒time curves of RAY1216 and TRA on linear scales truncated to 48 h after a single oral dose of 240 mL containing 400 mg/100 µCi [14C] RAY1216; ng eq/g, nanograms equivalent per gram; ng /ml, nanograms per millilitre.

Mean (±SE) [14C] RAY1216 concentrations (ng eq./g) in whole blood and plasma at each nominal time point ; Pharmacokinetic analysis set (all corresponding concentration‒time data within the collection window are included in the figure). SE, standard error of the mean.
Mass balance
Figure 5 also shows the mean cumulative excretion rate (% dose) in urine and feces samples from 0 to 192 h after a single fasting oral intake of 240 mL (containing 400 mg/100 μCi) [14C] RAY1216, with a collective and rapid increase in total radioactivity recovered in urine and feces from 0 to 48 h (approximately 65% within the first 48 h). Approximately 65% of the radiochemical dose accumulated and then gradually and slowly increased until the end, with an average of 104.17% of the total cumulative radioactivity excretion rate. None of the subjects experienced vomiting, and no vomitus was collected. The mean cumulative excretion rate (% dose) (mean + standard deviation) in urine and feces samples from 0 to 192 h after a single fasting oral intake of 240 mL (containing 400 mg/100 μCi) [14C] RAY1216.
Metabolite analysis and structural identification
Average radiogenic contribution to the human body of [14C] RAY1216 and its metabolites after oral administration (plasma (0–12 h), urine (0–48 h), feces (0–72 h)) and the mass-balanced population (N = 6).

apercentage of [14C]RAY1216 and its metabolites in the dose (% dose) = percentage of the total radioactive intensity , urine or fecal samples (% dose) × percentage of the total radioactive intensity , urine or fecal samples of [14C] RAY1216 and its metabolites in the total radioactive intensity/100.
b% AUC0–12 The ratio of total radioactivity in the region within the radioactive time range 0–12 h.
cUndetermined/Errors/Original form of the drug not included; MS: Detected only in mass spectrometry; NA: Not Applicable.
The results indicated that the parent compound RAY1216 was the main component in plasma, urine, and feces. [14C] RAY1216 accounted for 85.15% of the total radioactivity in the pooled plasma samples from 0 to 12 h, while metabolite M7 accounted for 7.37%. The cumulative radioactivity of other metabolites in plasma was less than 2%. The parent [14C] RAY1216 accounted for 23.85% of the total radioactivity in the pooled urine samples from 0 to 72 h, which was 1.22% of the administered dose. M186 and M168 accounted for 1.79% and 1.46%, respectively. M23 and M22 were the main metabolites in urine samples, accounting for 2.18% and 0.88% of the administered dose, respectively. The parent [14C] RAY1216 accounted for 47.15% of the administered dose in the pooled fecal samples from 0 to 120 h. M22 and M15 were the main metabolites in fecal samples, accounting for 17.29% and 11.10% of the administered dose, respectively. The proportion of other excreted metabolites was less than 10% of the administered dose. In addition, unlabelled metabolites of Oxidative dealkylation products were detected in plasma, urine and feces (where M25 and M26 were unlabelled metabolites of the parent drug part, while M23 was a metabolite of the labelled part). These metabolites were non-radioactive and could not be quantified by radioactivity. In summary, as shown in (Figure 6), in addition to the maternal drug RAY1216, eight metabolites were detected in plasma, urine and feces: Metabolites M22, M7 and M18 were Mono-oxidative forms of the maternal drug; Metabolite M15 is Hydrogenated form of the parent drug; Metabolites M23, M25 and M26 are produced by Oxidative dealkylation. Almost all metabolites are excreted in both urine and feces, with only M24 being detected in urine. Major metabolic pathways of [14C]RAY1216 in healthy subjects. Remarks: M25 and M26 are metabolites of the unlabelled portion of the original drug and have no radioactive intensity.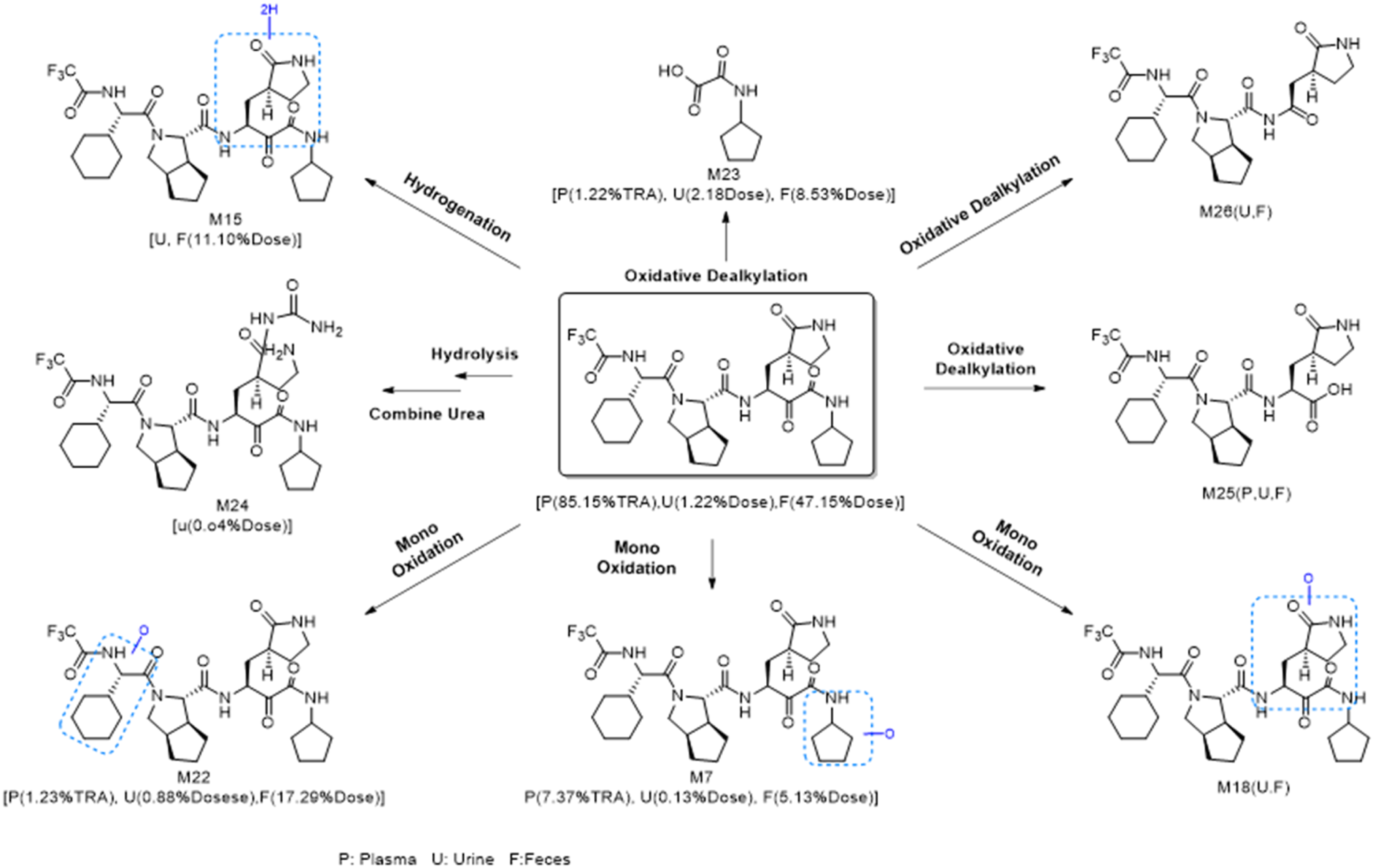
Safety results
Summary of adverse events (SS).

SOC: system organ class; PT: preferred term.
Discussion and conclusion
The present study involved a substance balance test of RAY1216 in healthy male subjects to evaluate the absorption, metabolism, and excretion characteristics of RAY1216 and its circulating metabolites in humans. The safety profile was good, with all adverse reactions being known from previous clinical trials. 14 The incidence of adverse reactions in the healthy cohort was significantly lower than that of therapeutic drugs such as Ensitrelvir 23 and Nirmatrelvir/Ritonavir. 24 No clinically significant abnormalities were observed in other safety-related examinations. The Phase III trial 25 and the validation of the PopPK (Population Pharmacokinetics) model 14 demonstrated that taking RAY1216 three times a day is sufficient to cover the replication cycle of SARS-CoV-2 (∼8–10 h).
After a single fasting oral administration of [14C] RAY1216 at 400 mg/100 µCi, the time to peak, half-life and Cmax of total plasma radioactivity were similar to those of the unlabelled parent drug of RAY1216, and the AUC0-t and AUC0-∞ were greater than those of the unlabelled parent drug, suggesting that most of the circulating radioactivity was composed mainly of the unlabelled parent drug of RAY1216. The total radioactivity concentration of whole blood was lower than that of plasma at all time points, and did not change with the change of plasma concentration, suggesting that [14C] RAY1216 preferentially binds to plasma rather than to blood cells. The average total cumulative excretion rate of radioactivity was 104.17%, the average total excretion (0–192 h) amounts in feces and urine accounted for 98.96% and 5.21% of the administered dose, respectively. which also indicated that RAY1216 was the major component (∼50%) in urine and feces. The radioconversion pathway indicates that there are multiple major pathways for the metabolism and elimination of RAY1216. In a non-clinical study (unpublished data), a single dose of [14C] RAY1216 (100 mg/100 µCi/kg, 0–168 h) in male rats resulted in cumulative excretion rates of 98.32% in total, 95.56% in urine and 2.76% in feces.
As a regulatory threshold for metabolites (<10% AUC), when the AUC of drug-related metabolites accounts for less than 10% of the total exposure of the parent drug (or all drug-related substances), it is generally not necessary to conduct additional non-clinical toxicology tests for the metabolite. If it is ≥10%, a dedicated safety evaluation must be initiated. More than 85% of the circulating radioactivity AUC in plasma was attributed to the parent RAY1216. Four metabolites circulated in plasma, with the average circulating radioactivity AUC of the metabolites being <7%. Therefore, this study is not expected to pose safety concerns that would necessitate additional toxicological evaluation. The measurement of circulating metabolite levels relative to total radioactivity is unlikely to raise safety concerns requiring further toxicological assessment, as these levels are <10% of the total drug-related exposure. Fewer uncharacterized metabolites were present in the plasma after the administration of [14C] RAY1216. This observation aligns with the results of the metabolite analysis.
The major metabolic pathways include Mono-oxidationM22 (18.17%), Hydrogenation M15 (11.10%), and Oxidative dealkylation M23 (10.72%). Non-clinical studies for the identification of metabolites were conducted in rats, cynomolgus monkeys, and beagle dogs. Notably, all metabolites identified in human hepatocytes were also detectable in at least one of the animal species examined (unpublished data).
Therefore, these metabolites were not considered to be toxicologically problematic In accordance with regulatory guidelines for metabolite safety testing. 26 These results suggest that the elimination of RAY1216 may predominantly occur through hepatic clearance or, at a minimum, via first-pass metabolism and biliary excretion. The significant increase in total radioactivity in the systemic circulation compared with that of the parent drug supports this potential effect of first-pass metabolism.
In addition, compared with the in vitro potency of RAY1216 (unpublished data), the Cmax in this study was more than 8 times the Effective Concentration 90%(EC90) in the Omicron model (the drug concentration required to achieve 90% max inhibition or effect), and the AUC0-∞ provided continuous exposure covering the viral replication cycle, indicating that the in vivo exposure of the single drug significantly exceeded the in vitro EC90 of all tested variants (≥4-fold safety margin), without the need for co-administration with Ritonavir. Compared with other approved Mpro inhibitors such as GST-HG171, Ensitrelvir, and Nirmatrelvir/Ritonavir, RAY1216 may have some advantages. RAY1216 and Nirmatrelvir/Ritonavir have the strongest activity, but RAY1216 maintains sub-120 nM activity against all variants and is less affected by key resistance mutations (such as E166A/L167F). 15 The t1/2 half-life of RAY1216 is better than that of GST-HG171 and Ensitrelvir, and the use of RAY1216 as a single drug may avoid the Drug-Drug Interaction (DDI)14,27 limitations of Nirmatrelvir/Ritonavir. Nirmatrelvir is mainly metabolized by CYP3A4, but its co-administration with Ritonavir leads to a significant increase in exposure, with Cmax > 3 µM approaching saturation, which may pose a moderate risk of enzyme saturation. In terms of safety, most antiviral drugs11,28,29 are mainly metabolized through renal pathways, while RAY1216 is a non-renal clearance dominant drug and does not require dose adjustment for patients with renal insufficiency, providing relatively safe medication for patients. 30 Compared with RAY1216, Bictegravir 28 and Obeldesivir 11 both have metabolite AUCs greater than 10%, requiring additional non-clinical toxicology data (repeated dosing, genotoxicity, target organ toxicity, etc.), and in subsequent clinical development, continuous monitoring of adverse events related to exposure-response is necessary, which may cause concerns among clinicians regarding the use of these drugs in special populations (such as patients with renal impairment).
The results of the preclinical study of the same drug 15 and the preclinical in vitro metabolic enzyme phenotype test in study (unpublished data) also revealed that CYP3A4 is the major metabolic enzyme. Therefore, the oral bioavailability of RAY1216 may be explained by the first-pass metabolism of CYP3A? This may explain the elevated liver enzyme levels and fewer SAEs with low renal creatinine observed by Bei Wang et al. 9 Moreover, the much lower excretion rate of urinary metabolites (4.44%) than the percentage of total radioactivity intensity (88.19%) may also be due to minimal low renal clearance (CL). CYP3A4 is a high-risk enzyme for drug interactions and is susceptible to strong inhibitors (such as azole antifungals) or inducers (such as antiepileptic drugs). Ketoconazole/voriconazole can increase the AUC of CYP3A4 substrates (such as ibrutinib) by 24–29 times, while carbamazepine/phenytoin induces CYP3A4, reducing itraconazole exposure by more than 50%, which may lead to treatment failure.31,32 Fecal excretion also suggests that when drugs that inhibit azole antifungals/P-gp (such as macrolides) are used in combination, azoles may inhibit P-gp (intestinal efflux pump), leading to the obstruction of the excretion of the original drug in bile, and cholestasis should be monitored. 33
This study investigated the ADME of RAY1216 in healthy individuals, but there may be expected differences in COVID-19 patients, those with liver function impairment, etc. Inflammatory factors such as IL-6 significantly reduce the expression of CYP3A4 by inhibiting nuclear receptors (such as PXR), leading to a decrease in CL and an increased risk of toxicity. The mRNA expression of efflux transporters such as P-gp can be reduced by up to 2 times under inflammatory stimulation, which may hinder the excretion of the original drug in bile or urine.34,35 Therefore, SARS-CoV-2 infection may affect ADME, and the above factors should be considered in subsequent studies. A study 25 also demonstrated that there were no clinically relevant differences in the exposure of RAY1216 in patients with liver function impairment. Patients with mild or moderate liver function impairment do not need to adjust the dose of RAY1216.
This study also has limitations. Although the median sample size of FDA-approved mass balance studies is 6, 36 the adverse reactions of COVID-19 treatment drugs and vaccines37–39 make us consider the possibility of rare adverse reactions of RAY1216. Moreover, due to concerns about the potential reproductive toxicity of radioactivity to women of childbearing age, only healthy men were allowed to participate in the study. However, due to the possibility of different expression levels of enzymes such as CYP1A2, CYP3A, and CYP4A1 between men and women,40,41 we have to consider the impact of gender differences on the study. Similarly, age differences are also a factor we need to consider whether RAY1216 can be a broad-spectrum drug). 42 In addition, due to regional restrictions, we only conducted the study on the East Asian population and did not consider inter-population differences. Therefore, conducting a more extensive future cohort study is very promising.
Conclusion
This research investigated the absorption, metabolism, and excretion of [14C] RAY1216 in healthy Chinese adult men show a good mass balance recovery. And clinical data were provided to help characterize the metabolic features of RAY1216 in the systemic circulation and excreta. The results indicated that [14C] RAY1216 was predominantly found in plasma in its parent drug, fecal are the main excretion routes of [14C] RAY1216. A higher proportion of fecal excretion indicates that the possibility of continuous monitoring for adverse events in subsequent clinical applications is relatively low, and clinicians have relatively less concerns about medication for special populations (such as patients with renal impairment), and the treatment has a broader scope. Therefore, the characteristic of ‘fecal dominance in excretion + low exposure to metabolites’ of RAY1216 provides a decisive basis for it to become a safe and low-interaction-risk oral treatment option for COVID-19 in patients with renal dysfunction.
Footnotes
Ethical approval
The questionnaire and methodology for this study (trial registration: NCT05523141) was approved by the Human Research Ethics committee of the University of Bengbu Medical University First Affiliated Hospital (Ethics approval number: 2023148).
Consent to participate
The study was performed in accordance with the ethical standards as laid down in the Declaration of Helsinki and its later amendments or comparable ethical standards. All subjects involved filled out the written informed consent form. All protocols, amendments, and consent forms were reviewed by the Institutional Review Board (IRB) or Ethics Committee (EC).
Consent for publication
The informed consent form has informed the subjects that all data will support the publication of research results, and research information will be legally authorized, but the confidentiality of personal information will not be violated.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: this work was supported by the (Key Project of Natural Science Research in Universities of Anhui Province, China); under Grant (number 2023AH040406); (General Project of Health Commission of Anhui Province, China) under Grant (number AHWJ2022b014); (Key Project of Natural Science Research in Universities of Anhui Province, China) under Grant (number 2024AH051141) and (Clinical Medical Research Transformation Project of Anhui Province, China) under Grant (number 202204295107020057).
Correction (October 2025):
Article updated to include Luning Sun and Huan Zhou in the “Corresponding authors” section.
Declaration of conflicting interests
The author(s) declared the following potential conflicts of interest with respect to the research, authorship, and/or publication of this article: The authors collectively affirm the absence of any actual or potential competing interests pertaining to this research. This declaration encompasses both tangible financial relationships and intangible academic influences that could be construed as affecting the objectivity of the presented findings.
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.
Clinical trial registration
https://www.clinicaltrials.gov/study/NCT06362460?intr=RAY1216&rank=3 identifier is NCT06362460.