
Abstract
Background
The COVID-19 pandemic has led to significant loss of life and economic disruption worldwide. Currently, there are limited effective treatments available for this disease. SARS-CoV-2 RNA-dependent RNA polymerase (SARS-CoV-2 RdRp) has been identified as a potential target for drug development against COVID-19. Natural products have been shown to possess antiviral properties, making them a promising source for developing drugs against SARS-CoV-2.
Objectives
The objective of this study is to identify the most effective natural inhibitors of SARS-CoV-2 RdRp among a set of 4924 African natural products using a multi-phase in silico approach.
Methods
The study utilized remdesivir (RTP), the co-crystallized ligand of RdRp, as a starting point to select compounds that have the most similar chemical structures among the examined set of compounds. Molecular fingerprints and structure similarity studies were carried out in the first part of the study. The second part of the study included molecular docking against SARS-CoV-2 RdRp (PDB ID: 7BV2) and Molecular Dynamics (MD) simulations including the calculation of RMSD, RMSF, Rg, SASA, hydrogen bonding, and PLIP. Moreover, the calculations of Molecular mechanics with generalised Born and surface area solvation (MM-GBSA) Lennard-Jones and Columbic electrostatic interaction energies have been conducted. Additionally, in silico ADMET and toxicity studies were performed to examine the drug likeness degrees of the selected compounds.
Results
Eight compounds were identified as the most effective natural inhibitors of SARS-CoV-2 RdRp. These compounds are kaempferol 3-galactoside, kaempferol 3-O-β-D-glucopyranoside, mangiferin methyl ether, luteolin 7-O-β-D-glucopyranoside, quercetin-O-β-D-3-glucopyranoside, 1-methoxy-3-indolylmethyl glucosinolate, naringenin, and asphodelin A 4’-O-β-D-glucopyranoside.
Conclusion
The results of this study provide valuable information for the development of natural product-based drugs against COVID-19. However, the elected compounds should be further studied in vitro and in vivo to confirm their efficacy in treating COVID-19.
Keywords
Introduction
As of May 9th, 2023, the World Health Organization (WHO) reported a total of 765,903,278 confirmed COVID-19 infections worldwide, along with 6,927,378 recorded deaths due to the virus. Even though the number of administrated vaccine doses reached 13,350,530,518, the fatal virus still shows an ability to affect vastly. 1 These alarming statistics underscore the significant effort required from scientists worldwide to discover a cure.
Over the past two decades, Computer-Aided Drug Discovery (CADD) approaches have gained significant traction in pharmacology development and testing.2,3 This widespread utilization can be attributed to several factors. Firstly, the elucidation of precise 3D structures of various protein targets within the human body. 4 Secondly, the remarkable advancements in computer software and hardware capabilities. 5 Lastly, the improved understanding of Structure–Activity Relationship (SAR) and Quantitative Structure–Activity Relationship (QSAR) principles. 6 Consequently, computer-aided chemistry approaches have been employed to investigate various pharmacokinetic and pharmacodynamics properties, enabling the identification of connections between chemical structure and activity. These methods include structure similarity analysis, 7 molecular fingerprints, 8 QSAR modeling, 9 pharmacophore analysis, 10 homology modeling, 11 molecular docking, 12 molecular dynamics (MD) simulations, 13 ADMET property predictions, 14 and density functional theory (DFT) calculations. 15
Throughout the annals of history, the nature has held an indispensable role in meeting fundamental human needs. Nature has not only provided solutions for medicinal treatments but has also offered sustenance and contributed to the formulation of various cosmetic products.16,17 Throughout the past 30 years, natural products have consistently contributed to nearly one-third of newly authorized pharmaceuticals by the FDA. For instance, the cytosine arabinoside, Cytarabine, which was used to treat acute myeloid leukaemia. 18 Eribulin, Halaven®, the anti-breast cancer, 19 the analgesic Ziconotide, Prialt®, 20 and the ergot alkaloid, dihydroergocristine, for Alzheimer's disease. 21 The continuity of this trend can be traced back to the 1940s, showcasing the enduring importance and worth of natural products within the realm of pharmaceutical research, development, and validation.
In targeting COVID-19, our team utilized various in silico methods to propose natural inhibitors. Four natural isoflavonoids were proposed as the most active inhibitors of hACE2 and viral main protease between fifty-nine isoflavonoids. 22 Likewise, fifteen alkaloidal compounds were studied in silico against five crucial proteins of COVID-19. 23 In a like manner, we designed multi-stage CADD approaches to recommend the most fitting inhibitor against certain COVID-19 enzymes. Among a set of 310 antiviral natural metabolites, potential inhibitors for nsp10, 24 the main protease,25,26 and the papain-like protease 27 have been determined. Furthermore, we investigated 3009 FDA-approved drugs and identified the most potent inhibitors against the SARS-CoV-2 nsp16-nsp10 2′-o-Methyltransferase Complex 28 and the RNA-Dependent RNA Polymerase. 29 Additionally, we explored a collection of 5956 compounds from traditional Chinese medicine to identify potential natural inhibitors for the SARS-CoV-2 Helicase. 30 Lastly, we discovered the most active semisynthetic inhibitor for the COVID-19 papain-like protease from a group of 69 molecules. 31
For this study, a curated set of 4924 African natural products was meticulously selected. 32 These compounds were isolated from diverse natural sources in Africa and obtained from the comprehensive African Natural Products Database (ANPDB). The dataset encompassed the period of 1962 to 2019, incorporating a wide range of scholarly sources such as international and local African journals, as well as MSc and Ph.D. theses archived in African university libraries. The selected compounds have been screened using different computational methods to detect the most potent SARS-CoV-2 RdRp inhibitors. Remdesivir, RTP, has been chosen as a lead compound due to its role as a co-crystallized ligand with the SARS-CoV-2 RdRp enzyme. Additionally, its robust potential to inhibit the SARS-CoV-2 RdRp has been demonstrated through both in silico computational predictions and in vitro experimental studies. 33 The utilized methods included molecular structure similarity and fingerprint studies against RTP, the molecular docking against SARS-CoV-2 RdRp, and several molecular dynamic (MD) simulation experiments.
Results and discussion
Filter using fingerprints
A co-crystallized ligand is a compound that exhibits a notable affinity for a specific protein, resulting in the formation of a crystallizable complex between the ligand and protein. 34 This complex offers valuable information about the interactions between the ligand and protein, unveiling crucial structural and chemical characteristics responsible for the strong binding affinity. The chemical structure of the co-crystallized ligand serves as a valuable blueprint for the design and development of inhibitors that can efficiently bind to the target protein, aiding in the discovery of potential therapeutic agents. 35 By analyzing the structural characteristics and functional groups of the co-crystallized ligand, we can develop a more profound comprehension of the critical elements responsible for its robust binding affinity. With this knowledge, we employed a strategic approach to select compounds exhibiting similar structural features to RTP. The objective was to identify potent inhibitors that display a high affinity for the RdRp protein. This selection process is rooted in the fundamental principle of Structure–Activity Relationship (SAR), which asserts that compounds with comparable chemical structures are likely to exhibit similar biological activities. 36
Most similar 200 candidates to RTP according to fingerprints similarity.

SA: The number bits in both RTP and the target candidates.
SB: The number bits in the target candidates but not RTP.
SC: The number bits in RTP but not the target candidates.
Molecular similarity
The base of the different molecular similarity studies is the principle that molecules of similar structures are predicted to exhibit similar biological activities.
43
The practical applications of molecular similarity as an essential approach in drug design have increased effectively in the last few years.
44
The molecular descriptors involved in this kind of study include the physicochemical properties and more specific features in the chemical structure such as H-bond donors (HBA),
45
H-bond acceptors (HBD),
46
the octanol–water partition coefficient (ALog p),
47
molecular weight (M. Wt),
48
rotatable bonds,
49
rings and aromatic rings,
50
besides molecular fractional polar surface area (MFPSA).
51
An investigation into molecular similarity, considering the aforementioned features, was conducted between the top 200 candidates showing the highest similarity (Figure 1) and RTP. This analysis was performed using Discovery Studio software to identify the most similar compounds. A subsequent filtration step by molecular docking was applied to a subset of thirty-four compounds (the selected most similar compounds). Similarity analysis between the tested candidates and RTP.
The software, Discovery Studio, was adjusted to compare the molecular descriptors that were mentioned before to select the most similar 40 candidates (Figure 2 and Table 2). Thirty-four compounds with good molecular similarity with the co-crystallized ligand (RTP) of RdRp (PDB ID: 7BV2). Most similar 34 candidates to RTP according to molecular similarity study.

Docking studies
Binding free energies (ΔG in Kcal/mol) of the studied candidates and


Superimposition of the co-crystallized pose (purple) and the re-docking pose (blue) of RTP indicate nearly the same binding mode.
RdRp enzyme consists of main three parts: (1) ATP binding site represented by a network of different amino acids including Asp760, Asn691, Ser759, Cys622, Lys545, and the key amino acid Arg555. (2) RNA primer is represented by many nucleotides including uridine 20 (U20), uridine 10 (U10), and adenine 11 (A11). (3) Pyrophosphate group (POP1003).
The mode of binding of RTP inside RdRp was illustrated in Figure 4. It was noticed that RTP interacted with the active site via the formation of four hydrogen bonds with U10 and U20, four hydrophobic interactions with U20 and A11 besides five electrostatic interactions with Arg555 and phosphate group. Three-dimension (A) and two-dimension (B) representations of the binding mode 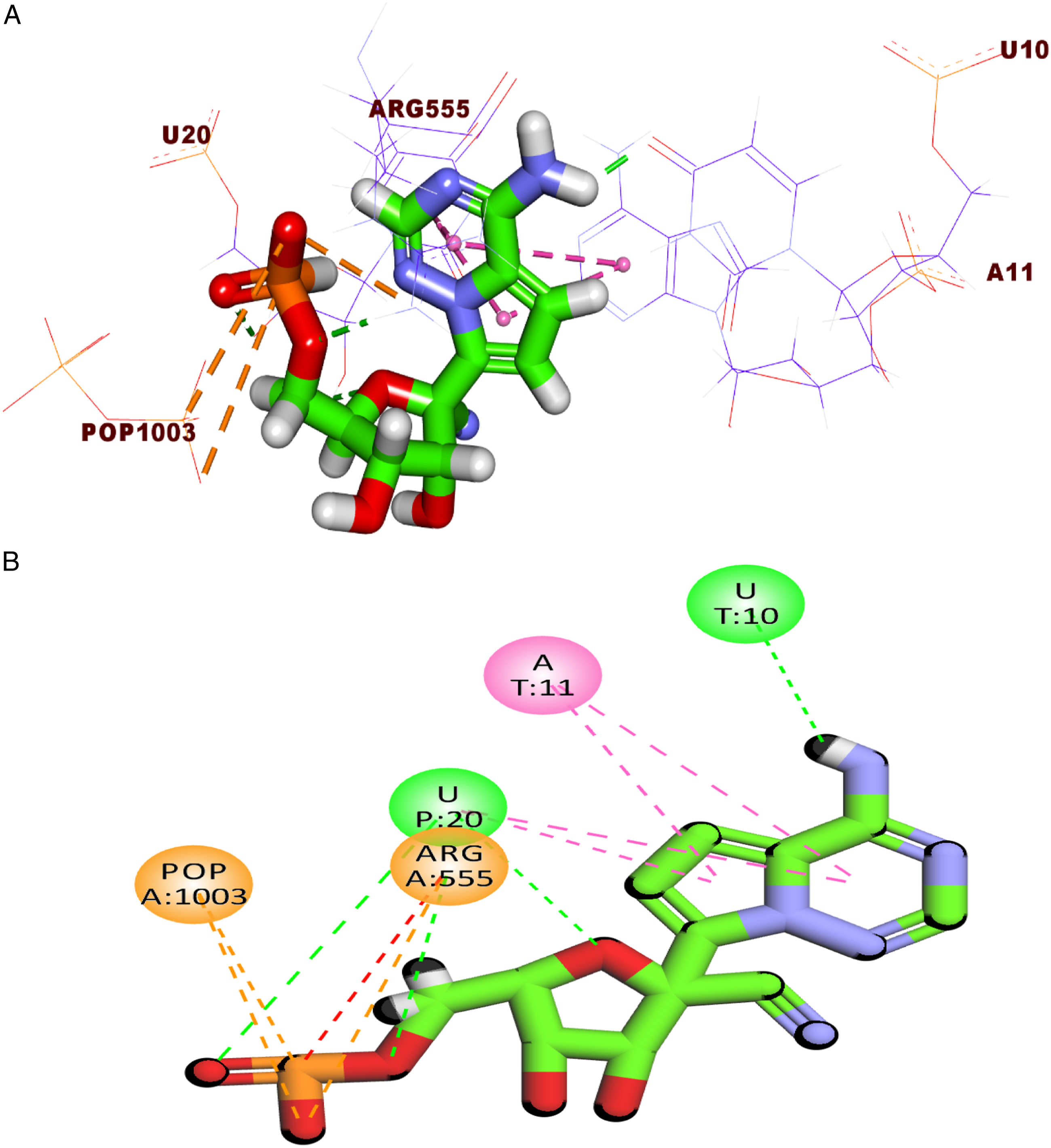
The binding of compound Mapping surface (A), three-dimension (B), and two-dimension (C) representations of compound 156 in the active site of RdRp. A: adenine, U: uracil, and POP: pyrophosphate.
Investigation of the top docking pose of compound Mapping surface, (A), three-dimension (B), and two-dimension (C) representations of compound 295 in the active site of RdRp. U: uracil and POP: pyrophosphate.
Next, the proposed binding pattern of the chromen-4-one derivative Mapping surface (A), three-dimension (B), and two-dimension (C) representations of compound 609 in the active site of RdRp. A: adenine, U: uracil, and POP: pyrophosphate.
The docking pose accomplished by compound 1123 (affinity value of −23.08) revealed that it occupied the RdRP active site. It interacted with the active site through seven hydrogen bonds with Asn691, Asp760, Arg555, and the pyrophosphate group. Likewise, the 1-methoxy-1H-indole moiety was involved in two electrostatic interactions with Arg555 besides three pi–pi interactions with U20 and A11 (Figure 8). Mapping surface (A), three-dimension (B), and two-dimension (C) representations of compound 
Finally, the metabolite Mapping surface, (A), three-dimension (B), and two-dimension (C) representations of compound 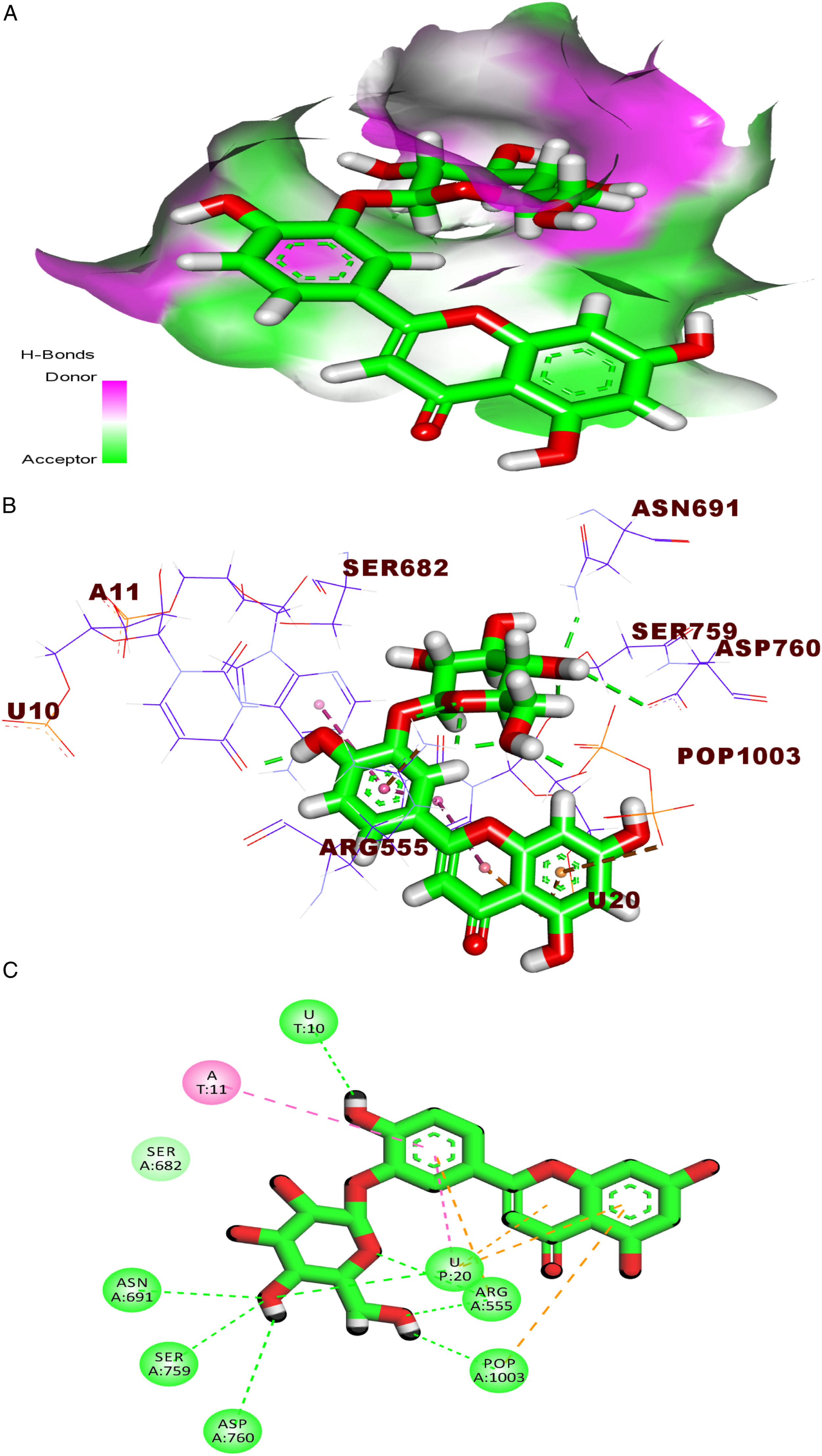
ADMET studies
Ten compounds with a higher energy score in the docking study were explored further utilizing computer-based ADMET experiments to determine their pharmacokinetic profiles. RTP was used as a reference. The calculated parameters include the potential to pass from the blood–brain barrier (BBB-L), the solubility level in aqueous media (S-L), the potential to be absorbed from the human gut (A-L) level, the potential to bind and inhibit the cytochrome P2D6 (CYP2D6-I), and the potential to bind to the plasma protein (PB-L).
Predicted ADMET descriptors for the examined compounds and RTP.

a0 is very high, 1 is high, 2 is medium, 3 is low, and 4 is very low.
b0 is good, 1 is moderate, 2 is poor, and 3 is very poor.
c0 is extremely low, 1 is very low, 2 is low, 3 is good, and 4 is optimal.
dN-I is a non-inhibitor and I is an inhibitor.
eL means less than 90% and M means more than 90%.
Toxicity studies
The toxicity of any new compound is a critical parameter in the process of drug development. The optimization of toxicity is the most time and money-consuming part of the pre-clinical trials during the drug discovery process. 52 Also, the accurate identification of the toxicological profile of a new compound is of great importance to avoid any side effects or late withdrawal. 53
Toxicity properties of tested compounds and the reference molecule.

aUnit: g/kg body.
Additionally, all members had rat maximum tolerated doses (RMTD-L) ranging from 0.577 to 1.574 g/kg body weight, which were extremely greater than RTP (0.003 g/kg body). Moreover, all compounds, except for
Furthermore
Molecular dynamic simulation
The MD simulation studies can explore several essential biomolecular processes that happen to a specific protein upon binding with a certain ligand over a determined time. These processes include protein conformational changes, protein folding, and stability of ligand binding. 54 All these processes have been predicted at an atomic level through the revelation of any change in the atomic positions at a temporal resolution of femtosecond. 55 Interestingly, such studies predict biomolecules’ responding, at an atomic level, to any perturbation such as mutation, protonation, phosphorylation, binding, or unbinding to a ligand.
To evaluate the integrity as well as the stability of the kaempferol 3-O-β-D-glucopyranoside-RdRp complex, the root-mean square deviation (RMSD) of the backbone atoms, root-mean square fluctuation (RMSF) of the Cα atoms, solvent accessible surface area (SASA), and the radius of gyration (Rg) of RdRp were investigated. In addition, the RMSD of the ligand, the number of H-bonds formed with the RdRp along the duration of the simulation, and the binding energy between the ligand and the protein using MM-GBSA were calculated (with decomposition).
The RMSD values of the protein which measures the overall change of each frame from the reference frame across the simulation show an increasing trend in the first half of the simulation and then stabilizes around 2.7 Å (Figure 10). This indicates a stable conformation during the second half of the simulation. The RMSF values show a large fluctuation from Arg331 to Asn360, which corresponds to the loop motion during the simulation (Figure 11). The SASA values (Figure 12) identify how much of the protein surface is exposed to the solvent, and the radius of gyration (Rg) (Figure 13) measures the protein compactness. Both of them indicate stable values around 37,300 Å2 and 28.8 Å for the SASA and Rg, respectively, indicating that the RdRp was stably folded, which was critical for the validity of the simulation analysis. RMSD of the protein in RdRp-kaempferol 3-O-β-D-glucopyranoside complex, calculated throughout a 100-ns MD simulation. RMSF of RdRp Cα atoms when complexed with kaempferol 3-O-β-D-glucopyranoside, calculated over the course of a 100-ns MD simulation. SASA values of RdRp when complexed with kaempferol 3-O-β-D-glucopyranoside, calculated for the 100-ns MD simulation. Radius of gyration of RdRp when complexed with kaempferol 3-O-β-D-glucopyranoside, calculated over the course of a 100-ns MD simulation.



Furthermore, the RMSD of kaempferol 3-O-β-D-glucopyranoside is a measure of how much its conformation has varied relative to the reference structure. kaempferol 3-O-β-D-glucopyranoside reached a relatively stable conformation after 25 ns, with an RMSD fluctuating around 0.48 nm (Figure 14), indicating small changes in the kaempferol 3-O-β-D-glucopyranoside’s position and conformation while preserving its binding to the RdRp’s active site. Additionally, the number of H-bonds formed between the compound and RdRp was calculated (Figure 15) and was shown to have two to three bonds, on average. RMSD of the ligand with reference to the starting configuration, calculated over the course of the 100-ns simulation. Change in the number of H-bonds formed between the ligand and the RdRp over the course of the 100-ns simulation.

Additionally, the RdRp-kaempferol 3-O-β-D-glucopyranoside interactions were analyzed to assess the kaempferol 3-O-β-D-glucopyranoside’s affinity towards the RdRp and the strength of their interactions. The energetics analysis revealed that the average Lennard-Jones energy was −92.5382 kJ/mol (−22.11 kcal/mol) (Figure 16), and the average Coulombic interaction energy was −206.63 kJ/mol (−49.42 kcal/mol) (Figure 17), indicating the ligand’s high affinity towards the RdRp. Lennard-Jones interaction energy during the MD simulation, showing an average value of −92.5382 kJ/mol. Columbic electrostatic interaction energy during MD simulation, showing an average value of −206.63 kJ/mol.

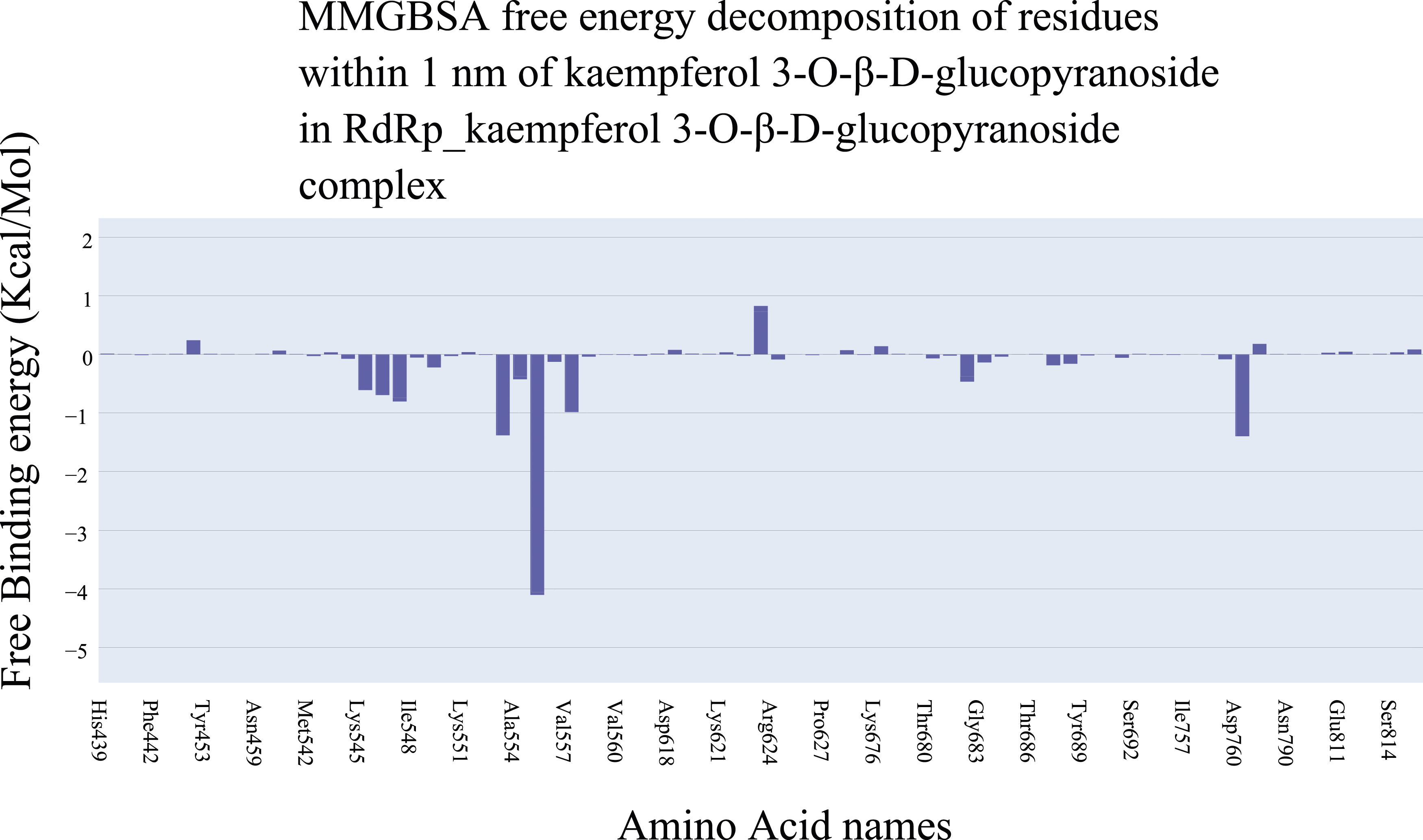
Moreover, the RdRp _kaempferol 3-O-β-D-glucopyranoside interactions were analyzed to assess the kaempferol 3-O-β-D-glucopyranoside’s affinity towards the RdRp and the contribution of amino acids around 1 nm of the compound. The binding energy analysis using MM-GBSA revealed that the average binding energy is −25 kcal/mol (Figure 18) with electrostatic interactions as the most effective component (−67.33 Kcal/Mol) compared to the hydrophobic interactions (−25.63 Kcal/Mol). In addition, decomposition was performed to obtain the contribution of each amino acid towards the binding energy (Figure 19). There are three amino acids (Arg553 [−1.38 Kcal/Mol], Arg555 [−4.1 Kcal/Mol], and Asp760 [−1.39 Kcal/Mol]) with a contribution of less than −1 Kcal/Mol which are consistent with the docking and PLIP results. Different energetic components of MM-GBSA and their values. Bars represent the standard deviations. Binding free energy decomposition of the RdRp_kaempferol 3-O-β-D-glucopyranoside complex.
Protein–ligand interaction profiler experiment
PLIP stands as a valuable computational tool employed to meticulously dissect and visualize the intricate interactions between a protein and a ligand, those diminutive molecules of significance. 56 This tool affords a closer, atomic-level look into the profound engagement of a ligand with a protein. Its prowess lies in unraveling binding sites, exposing the nuances of hydrogen bonding, shedding light on hydrophobic interactions, and unearthing an array of pertinent information. Such insights are tantamount to unraveling the complex behavior exhibited by the intricate interplay of protein–ligand complexes. 57
Interacting amino acids obtained from PLIP for each representative cluster. Bold amino acids are the amino acids that are common between the docking step and the MD simulation.


Clustering of the trajectory using TTClust and the interactions obtained from PLIP on the representative frames.
Conclusion
Eight compounds among 4924 African natural products (kaempferol 3-galactoside, kaempferol 3-O-β-D-glucopyranoside, mangiferin methyl ether, luteolin 7-O-β-4C1-D-glucopyranoside, quercetin-3-glucopyranoside, 1-methoxy-3-indolylmethyl glucosinolate, naringenin, and asphodelin A 4’-O-β-D-glucopyranoside) were decided as the most potent inhibitors against SARS-CoV-2 RdRp (PDB ID: 7BV2). The study depended on a multi-phase in silico approach to select the most structurally similar compounds to RTP, the co-crystallized ligand of the RdRp employing a molecular fingerprints and structure similarity studies. Then, molecular docking for the chosen 34 compounds preferred 10 candidates. Followingly, ADMET study excluded a compound. Finally, MD simulation studies verified the correct binding of kaempferol 3-O-β-D-glucopyranoside against RdRp for 100 ns. The outputted results are a solid base to conduct further in vitro and in vivo studies on the preferred compounds to find a cure against COVID-19.
Method
Fingerprint studies
For the fingerprints analysis of 4924 African natural products to select the most similar 200 compounds to RTP, we utilized the Discovery Studio 4.0 software to compare the number of charges, H. bond acceptors, H. bond donors, f-positive ionizable atoms, negative ionizable atoms, halogen atoms, and aromatic rings in addition to hybridization and ALogP as described in the Supplementary data section.
Molecular similarity detection
For the molecular similarity analysis of 200 African natural products, to select the most similar compounds (34 compounds) to RTP, we utilized the Discovery Studio 4.0 software to compare the number of rotatable bonds, rings, aromatic rings, H. bond donors, and H. bond acceptors, in addition to ALog p, molecular weight, and the MFPSA as described in the Supplementary data section. A maximum distance of 1.385 was selected.
Docking studies
Docking studies were performed against SARS-CoV-2 RdRp (PDB ID: 7BV2) using Discovery Studio software 58 and depending on RTP as a reference (see method part in Supplementary data).
ADMET analysis
Discovery Studio 4.0 was used to predict human intestinal absorption, aqueous solubility, blood–brain barrier penetration, plasma protein binding, cytochrome P450 2D6 inhibition, and hepatotoxicity (see method part in Supplementary data).59,60
Toxicity studies
Discovery Studio 4.0 software was used 61 to predict the FDA rat carcinogenicity, TD50, MTD, oral LD50 in rats, LOAEL, ocular, and dermal irritancy (see method part in Supplementary data).
Molecular dynamics simulations
A 100 ns molecular dynamics (MD) simulation was performed for the kaempferol 3-O-β-D-glucopyranoside–RdR complex using GROMACS v.2021.4. Once the simulation concluded, the periodic boundary conditions (PBCs) were eliminated from the system using the trjconv command, restoring the RdRp protein to its complete form. Subsequently, VMD TK scripts were applied to analyze the trajectory. Calculations were conducted for the root-mean square deviation (RMSD) of both RdRp and kaempferol 3-O-β-D-glucopyranoside. Essential metrics such as hydrogen bond count, radius of gyration (RoG), solvent-accessible surface area (SASA), and root-mean square fluctuation (RMSF) were also determined. Following this, the trajectory of the kaempferol 3-O-β-D-glucopyranoside-RdRp complex underwent clustering through TTClust, generating representative frames for each cluster. To comprehensively examine these frames for the spectrum and types of kaempferol 3-O-β-D-glucopyranoside–RdRp interactions, the Protein–Ligand Interaction Profiler (PLIP) was enlisted. The outcomes were compiled in a .pse file format, readily viewable through PyMOL. In order to ascertain the binding free energy, the MM-GBSA method was employed using the gmx_MMPBSA programme. Furthermore, decomposition analysis was executed to quantify the contributions of interacting amino acids within 1 nm of kaempferol 3-O-β-D-glucopyranoside. Notably, the ionic strength was set at 0.154 M, and the solvation method (igb) was designated as 5 for the calculations (see method part in Supplementary data).
Supplemental Material
Supplemental Material - Computer-assisted drug discovery of potential natural inhibitors of the SARS-CoV-2 RNA-dependent RNA polymerase through a multi-phase in silico approach
Supplemental Material for Computer-assisted drug discovery of potential natural inhibitors of the SARS-CoV-2 RNA-dependent RNA polymerase through a multi-phase in silico approach by Eslam B Elkaeed, Bshra A Alsfouk, Tuqa H Ibrahim, Reem K Arafa, Hazem Elkady, Ibrahim M Ibrahim, Ibrahim H Eissa, and Ahmed M Metwaly in Antiviral Therapy.
Footnotes
Author contributions
The study was conceptualized, designed, and supervised by IHE and AMM. EE and HE carried out the computational studies. TI and RA carried out the MD studies. HE was responsible for analyzing and interpreting the data. The fund accusation was provided by EBE and BAA, who were also included in the writing of the manuscript. All authors have thoroughly reviewed and agreed upon the final version of the manuscript.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This research was funded by Princess Nourah bint Abdulrahman University Researchers Supporting Project number (PNURSP2023R142), Princess Nourah bint Abdulrahman University, Riyadh, Saudi Arabia. The authors extend their appreciation to the Research Center at AlMaarefa University for funding this work.
Supplemental Material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.