
Abstract
Objective
Medicinal herbs are being investigated for medicationhg development against SARS-CoV-2 as a rich source of bioactive chemicals. One of the finest approaches for finding therapeutically effective drug molecules in real time is virtual screening scheme such as molecular docking in conjunction with molecular dynamics (MD) simulation. These virtual techniques provide an ample opportunity for the screening of plausible inhibitors of SARS-CoV-2 different target proteins from a comprehensive and extensive phytochemical library. The study was designed to identify potential phytochemicals by virtual screening against different receptor proteins.
Methods
In the current study, a library of plant secondary metabolites was created by manually curating 120 phytochemicals known to have antimicrobial as well as antiviral properties. In the current study, different potential phytochemicals were identified by virtual screening against various selected receptor proteins (i.e., viral main proteases, RNA-dependent RNA polymerase (RdRp), ADP ribose phosphatase, nonstructural proteins NSP7, NSP8, and NSP9) which are key proteins responsible for transcription, replication and maturation of SARS-CoV-2 in the host. Top three phytochemicals were selected against each viral receptor protein based on their best S-scores, RMSD values, molecular interactions, binding patterns and drug-likeness properties.
Results
The results of molecular docking study revealed that phytochemicals (i.e., baicalin, betaxanthin, epigallocatechin, fomecin A, gallic acid, hortensin, ichangin, kaempferol, limonoic acid, myricetin hexaacetat, pedalitin, quercetin, quercitrin, and silvestrol) have strong antiviral potential against SARS-CoV-2. Additionally, the reported preeminent reliable phytochemicals also revealed toxicity by no means during the evaluation through ADMET profiling. Moreover, the MD simulation study also exhibited thermal stability and stable binding affinity of the pedalitin with SARS-CoV-2 RdRp and SARS-CoV-2 main protease which suggests appreciable efficacy of the lead optimization.
Conclusion
The biological activity and pharmacologically distinguishing characteristics of these lead compounds also satisfied as repurposing antiviral drug contenders and are worth substantial evaluation in the biological laboratory for the recommendation of being plausible antiviral drug candidates against SARS-CoV-2.
Keywords
Introduction
The novel coronavirus disease (COVID-19) is caused by the single-stranded RNA virus, the severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2), which is typified by shortness of breath, cough, and fever with additional symptoms such as muscle aches, respiratory tract illness, severe pneumonia, loss of smell, weariness, and death.1,2 As a result of its high human-to-human contagiousness, the virus expanded to 187 countries and territories and infected 271,963,258 people as of 17 December 2021, with a death count of 5,331,019 (https://covid19.who.int/). On 30 January 2020, the World Health Organization (WHO) proclaimed the SARS-CoV-2 outbreak a public health emergency of international concern (PHEIC) and a pandemic on 11 March 2020. 3
Currently, there is no effective treatment for the novel SARS-CoV-2 virus. As a result, efforts were made to recognize infected people through quick diagnosis and then isolate them to prevent the disease from spreading further. 3 Other advised actions to control the spread of this virus include wearing masks, washing hands with soap, and maintaining social distance. To prevent COVID-19 infection, authorized pharmaceuticals, therapies in clinical trials, and substances derived from medicinal plant extracts are all being researched at the same time.4,5 Currently, scientists from several countries are working to find a vaccine against the virus. However, developing vaccines for RNA viruses is a challenging task as seen by the cases of HIV, influenza, and hepatitis C, among others.
Traditional remedies are primarily made up of natural phytochemicals. They constitute, without a doubt, a significant source of essential chemicals needed for the development of novel pharmaceutical medicines; however, because of practical difficulties in investigating them in molecular target specific tests, their utilization has diminished in the previous 25 years. 6 With the development of new sophisticated methodologies, these technical impediments to natural phytochemical screening are now being reduced. 7 The medicinal value of a phytochemical is directly proportional to the mechanism through which it interacts with its cell target. It is the most rational and significant step in the drug discovery process. There are a number of pharmacological targets to block in the therapy of retroviruses (e.g., HIV), including the viral envelope protein, RNA, reverse transcriptase, integrase, and major protease enzyme.5,8
In silico target identification for a phytochemical is a cost-effective, time-efficient, and beneficial virtual screening approach. 9 Quantitative structure activity relationship (QSAR) and molecular docking are the two most extensively used methods in silico approaches. 10 For synthetic compounds and phytochemicals, the last one can be used to quickly identify target biomacromolecules (DNA, RNA, and proteins). The method also aids in the understanding of natural compounds' mechanisms of action with their target proteins, as well as the development of new derived medications. 9 Several phytochemical-based medicines have been developed via protein-ligand docking. 5
The SARS-CoV-2 main proteases (Mpro) play a role in the cleavage of long polypeptides into smaller pieces to make them functional. 11 RNA-dependent RNA polymerase (RdRp) is involved in RNA synthesis, transcription, and replication process along with nonstructural proteins NSP7 and NSP8. 5 ADP ribose phosphatase of NSP3 and NSP9 RNA binding protein are the key receptors for the development of new therapeutic drug targets. 12 The ongoing COVID-19 outbreak, which is linked to a highly pathogenic variant of the nCoV-19 virus, has underlined the urgent need for medication research. COVID-19 infection necessitates nucleotide analogue-based antiviral medicines that target the exterior components of the RNA virus’s reproduction and functional machinery. 13 Therefore, the current research work was aimed to limit viral transcription, replication, and maturation by targeting Mpro, RdRp, and other critical nonstructural proteins with various plant-based phytochemicals. The purpose of this study was to target various viral proteins at one platform which are essential for viral replication, transcription, and translation. Moreover, the comparison of different interactions between ligand-receptor complexes would help researchers to locate more precise target site in the receptor proteins. The targeting of multiple viral proteins with active residues at one time can display a single leading drug candidate against multiple proteins which can provide a great help in controlling the COVID-19 infection in a timely manner. It will navigate the path towards the exploration of more potential drug candidates with extensive bioavailability and efficacy against SARS-CoV-2 infection.
Materials and methods
Ligand preparation
A comprehensive literature screening was done in order to screen the most effective antiviral bioactive compounds counter to variety of antiviral infections. The chemical structures of 120 phytochemicals were downloaded from the PubChem database in SDF format. 14 A ready-to-dock library of 120 ligands was prepared after energy minimization with default parameters using Molecular Operating Environment (MOE). 15 Along with 120 ligands, two antiviral agents (i.e., remdesivir and nirmatrelvir) were downloaded and used as control drug molecules. For COVID-19 patients, these two drugs have been recommended by the FDA.
Retrieval and optimization of receptor proteins
For this study, the 3D-structures of different SARS-CoV-2 (COVID-19) receptor proteins including SARS-CoV-2 RNA-dependent RNA polymerase (RdRp) [PDB ID: 6M71], 16 ADP ribose phosphatase of NSP3 [PDB ID: 6VXS], 17 NSP9 RNA binding protein [PDB ID: 6W4B], 18 NSP7 and NSP8 complex [PDB ID: 7DCD], 19 SARS-CoV-2 main protease (Mpro) [PDB ID: 7L0D], 20 and SARS-CoV-2 main protease C145S mutant [PDB ID: 7N5Z] 21 were downloaded from Protein Data Bank in .pdb format. 22 The selected receptor proteins were refined and optimized using MOE software with default parameters. Optimization was done by removing ligands and water molecules, addition of H-atoms, charge adjustments, energy minimization, and 3D protonation with force field gradient: 0.05.
Molecular docking
Molecular docking was performed after finding the active site residues of receptor proteins through site finder tool of MOE software. A library of ready-to-dock ligands was docked against active site residues of the binding pocket of selected receptor proteins and top three candidates against each receptor protein were selected on the basis of their best S-scores and binding patterns between ligands and targeted proteins.
Drug scanning
Lipinski’s rule of five was executed to identify drug-like or non-drug-like behavior of the selected compounds. 23 It predicts the success or failure of drug candidates in the preclinical stage. According to these rules, the molecular mass of the compounds should be < 500, hydrogen bond donors should be ≤ 5, hydrogen bond acceptors should be ≤ 10, logP value should be ≤ 5, and molecular refractive index should between 40 and 130. 24
ADMET profiling
The online tool admetSAR 2.0 25 (http://lmmd.ecust.edu.cn/admetsar2/) was used for ADMET profiling of potential drug candidates. ADMET classify drugs in five aspects (absorption, distribution, metabolism, excretion, and toxicity) to recognize the drug bioavailability from chemical biochemistry perspective. Only those compounds which accomplish all the pharmokinetics parameters could further be reported as potential drug candidates against selected viral receptor proteins.
Molecular dynamics (MD) simulation
For 100 nanoseconds, Desmond, a software from Schrödinger LLC, was used to model molecular dynamics. 26 The earliest phase of receptor and ligand complexes for molecular dynamics simulation is always the docking experiments. MD studies can predict ligand binding state in static situations and docking helps to provide a static view of the binding pose of a molecule at the active site of the protein. 27 By integrating Newton’s classical equation of motion, MD simulations typically compute atom movements over time. Simulations were used in this study to predict the ligand binding status in the physiological environment.28,29
Based on the results of the molecular docking, druggability, and ADMET profiling the phytochemical pedalitin was selected for MD simulation against two receptor proteins (i.e., SARS-CoV-2 RdRp and SARS-CoV-2 main protease). The quality of both protein structures was evaluated using PROCHECK Ramachandran plots.
30
The receptor–ligand complex was preprocessed using Protein Preparation Wizard of Maestro, which included complex optimization and minimization. All of the systems were prepared using the System Builder tool. TIP3P, a solvent model with an orthorhombic box, was chosen (Transferable Intermolecular Interaction Potential 3 Points). In the simulation, the OPLS 2005 force field was used.
31
To make the models neutral, counter ions were introduced. To mimic physiological conditions, 0.15
Results
Molecular docking analysis
Molecular docking scores, root mean square deviation values, and interactions of selected phytochemicals against six receptor proteins from SARS-CoV-2.

Remdesivir
Nirmatrelvir.
The results presented in Table 1 clearly show that all the compounds bind to selected viral proteins to varying degrees. Different ligands have the highest binding affinities for different proteins and the best protein-ligand interaction for each protein is shown in Figures 1–6. Pedalitin has binding energy of −13.832 kcal/mol against SARS-CoV-2 RdRp and form hydrogen bonds with Thr556 and Tyr619 amino acid residues of the receptor protein (Figure 1). In case of ADP ribose phosphatase of NSP3 from SARS-CoV-2, the compound myricetin hexaacetate exhibited the lowest binding energy of −12.990 kcal/mol and molecular interactions for hydrogen bonding involved LysA90 of the receptor protein (Figure 2). Binding interactions of pedalitin with SARS-CoV-2 RNA-dependent RNA polymerase revealed through protein-ligand approach. (a) Interactions of pedalitin with the receptor; (b) Binding patterns of pedalitin with the receptor protein. Binding interactions of myricetin hexaacetate with ADP ribose phosphatase of NSP3 from SARS-CoV-2 revealed through protein-ligand approach. (a) Interactions of myricetin hexaacetate with the receptor; (b) Binding patterns of myricetin hexaacetate with the receptor protein. Binding interactions of limonoic acid with NSP9 RNA binding protein of SARS-CoV-2 revealed through protein-ligand approach. (a) Interactions of limonoic acid with the receptor; (b) Binding patterns of limonoic acid with the receptor protein. Binding interactions of betaxanthin with NSP7 and NSP8 complex of SARS-CoV-2 revealed through protein-ligand approach. (a) Interactions of betaxanthin with the receptor; (b) Binding patterns of betaxanthin with the receptor protein. Binding interactions of quercitrin with SARS-CoV-2 main protease (Mpro) revealed through protein-ligand approach. (a) Interactions of quercitrin with the receptor; (b) Binding patterns of quercitrin with the receptor protein. Binding interactions of fomecin A with SARS-CoV-2 main protease C145S mutant with SARS-CoV-2 main protease (Mpro) revealed through protein-ligand approach. (a) Interactions of fomecin A with the receptor; (b) Binding patterns of fomecin A with the receptor protein.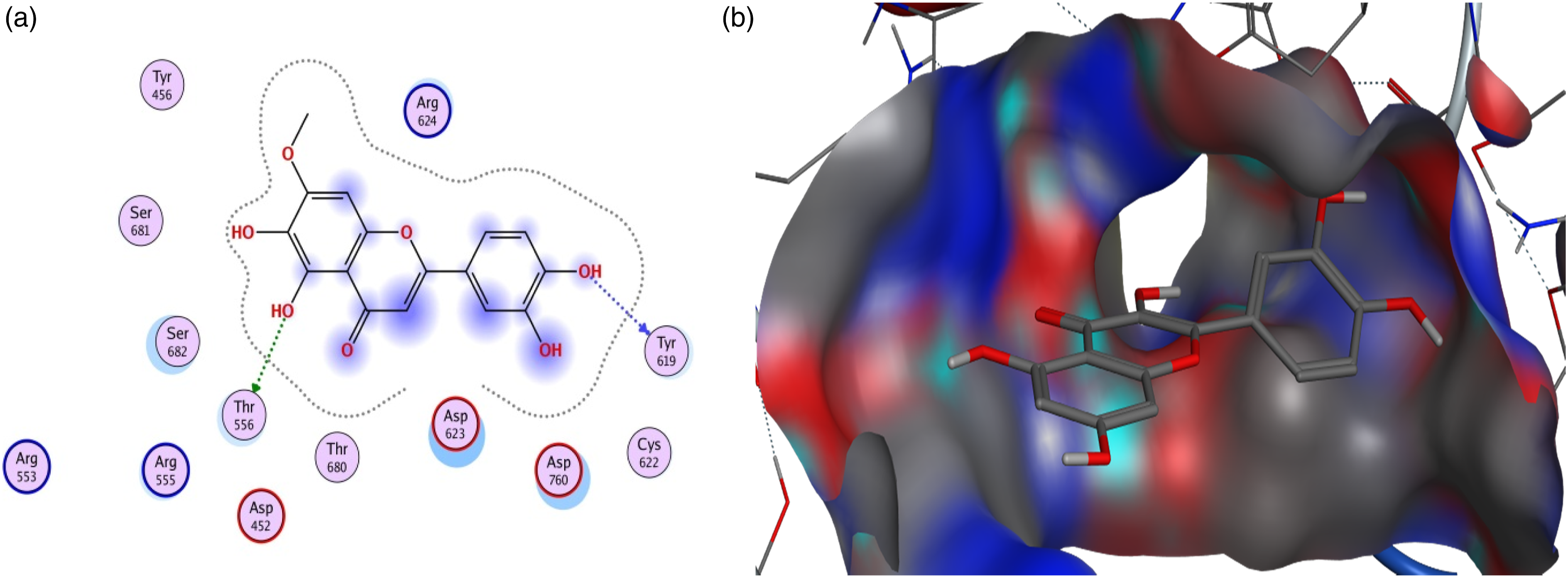
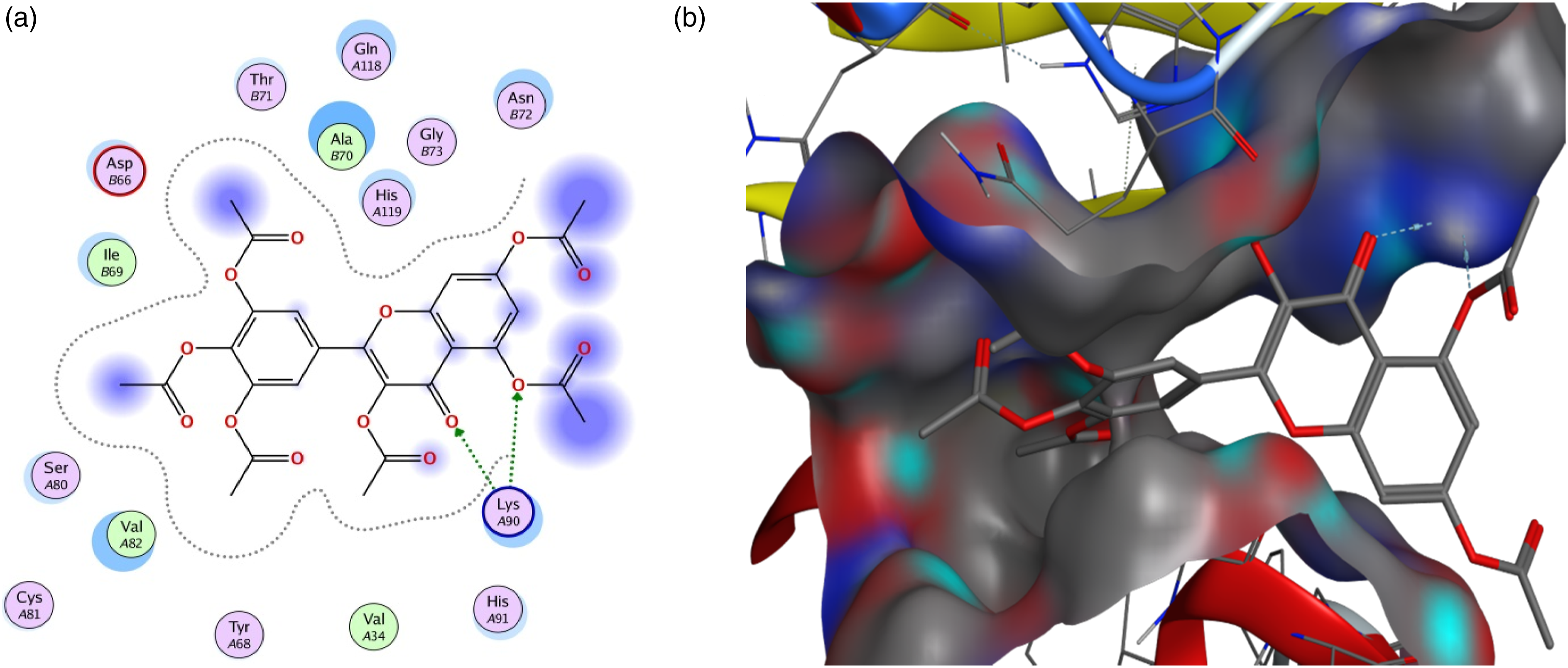



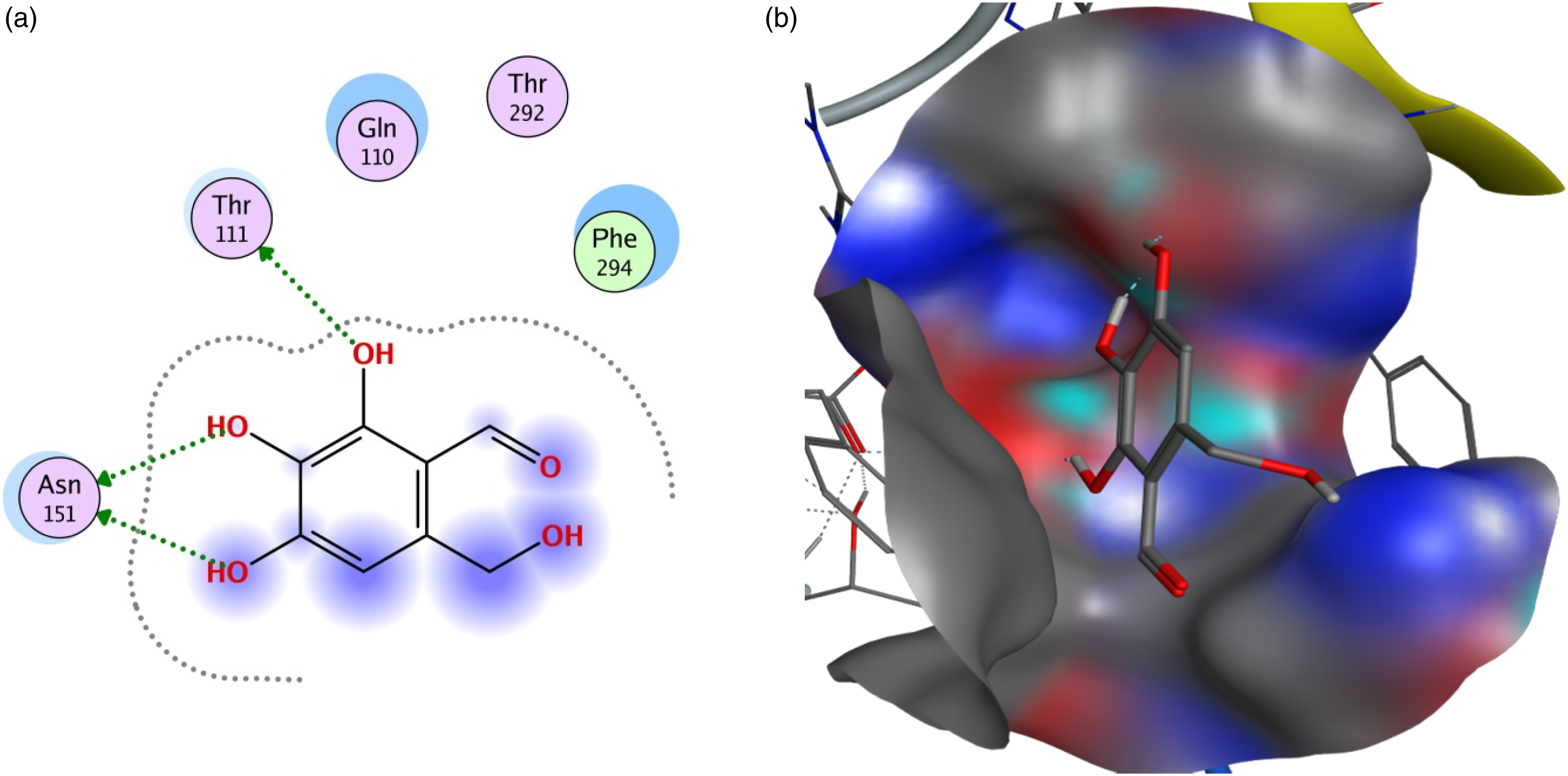
The Nsp9 RNA binding protein of SARS-CoV-2 showed lowest binding energy with limonoic acid (−12.271 kcal/mol), and CysA74, SerB6, and ValB8 residues of the receptor protein was involved in the molecular interactions and binding patterns (Figure 3). Betaxanthin revealed −12.946 kcal/mol energy against NSP7 and NSP8 complex of SARS-CoV-2 and showed binding interactions with LysA127, and LysC127 residues of the receptor protein (Figure 4). For SARS-CoV-2 Mpro, quercitrin showed −13.35 kcal/mol binding energy and exhibited interactions with Thr26, His163, and Glu166 residues of the receptor protein (Figure 5). Fomecin A with binding energy of −12.325 kcal/mol showed strong interactions with SARS-CoV-2 main protease from C145S mutant and the interactions involved Thr111 and Asn151 residues in the docking site of the receptor protein for hydrogen bond formations (Figure 6).
The two FDA-approved drugs (i.e., remdesivir and nirmatrelvir, as anti-SARS-CoV-2 drugs) were used as control to compare with best ligands found in this study. The results of molecular docking of 120 ligands against six receptor proteins displayed that the phytochemical pedalitin revealed maximum efficacy and binding interaction with best S-score against two viral proteins (i.e., RdRp and mutant main protease C145S) of SARS-CoV-2. The controls were also docked against both receptor proteins and the results were compared in order to find out the efficacy of pedalitin over remdesivir and nirmatrelvir to support the findings of this study. The results showed that for RdRp of SARS-CoV-2, remdesivir (S-score: −8.0884 kcal/mol) (Figure S1) and nirmatrelvir (S-score: −5.9458 kcal/mol) (Figure S2) interacted with Arg555, Asp760, and Thr556 residues of the active sites and Arg555 was found as a common interacting residue. The interactions and binding patterns of control drugs (i.e., remdesivir and nirmatrelvir) with other selected receptor proteins (i.e., ADP ribose phosphatase of NSP3, Nsp9 RNA binding protein of SARS-CoV-2, Nsp7 and 8 complex of SARS-CoV-2, and SARS-CoV-2 main protease) have been shown in Figures S3–S10. The control drugs remdesivir (S-score: −6.7179 kcal/mol) (Figure S11) and nirmatrelvir (S-score: −6.5849 kcal/mol) (Figure S12) interacted with common amino acid Asp153 of the mutant main protease C145S of SARS-CoV-2. The results of this study exhibited better performance of pedalitin as potential anti-SARS-CoV-2 drug against all selected receptor proteins.
Drug scanning and ADMET profiling
Evaluation of selected ligands through Lipinski’s rule of five.

6M71: SARS-CoV-2 RNA-dependent RNA polymerase (RdRp), 6VXS: ADP ribose phosphatase of NSP3, 6W4B: NSP9 RNA binding protein, 7DCD: NSP7 and NSP8 complex, 7L0D: SARS-CoV-2 main protease (Mpro), 7N5Z: SARS-CoV-2 main protease C145S mutant. MW: Molecular weight, HBD: Number of hydrogen bond donors; HBA: Number of hydrogen bond acceptors, logP: the logarithm of octanol/water partition coefficient, A: Molar refractivity, nrotb: Number of rotatable bonds.
ADMET profiling of selected ligands.
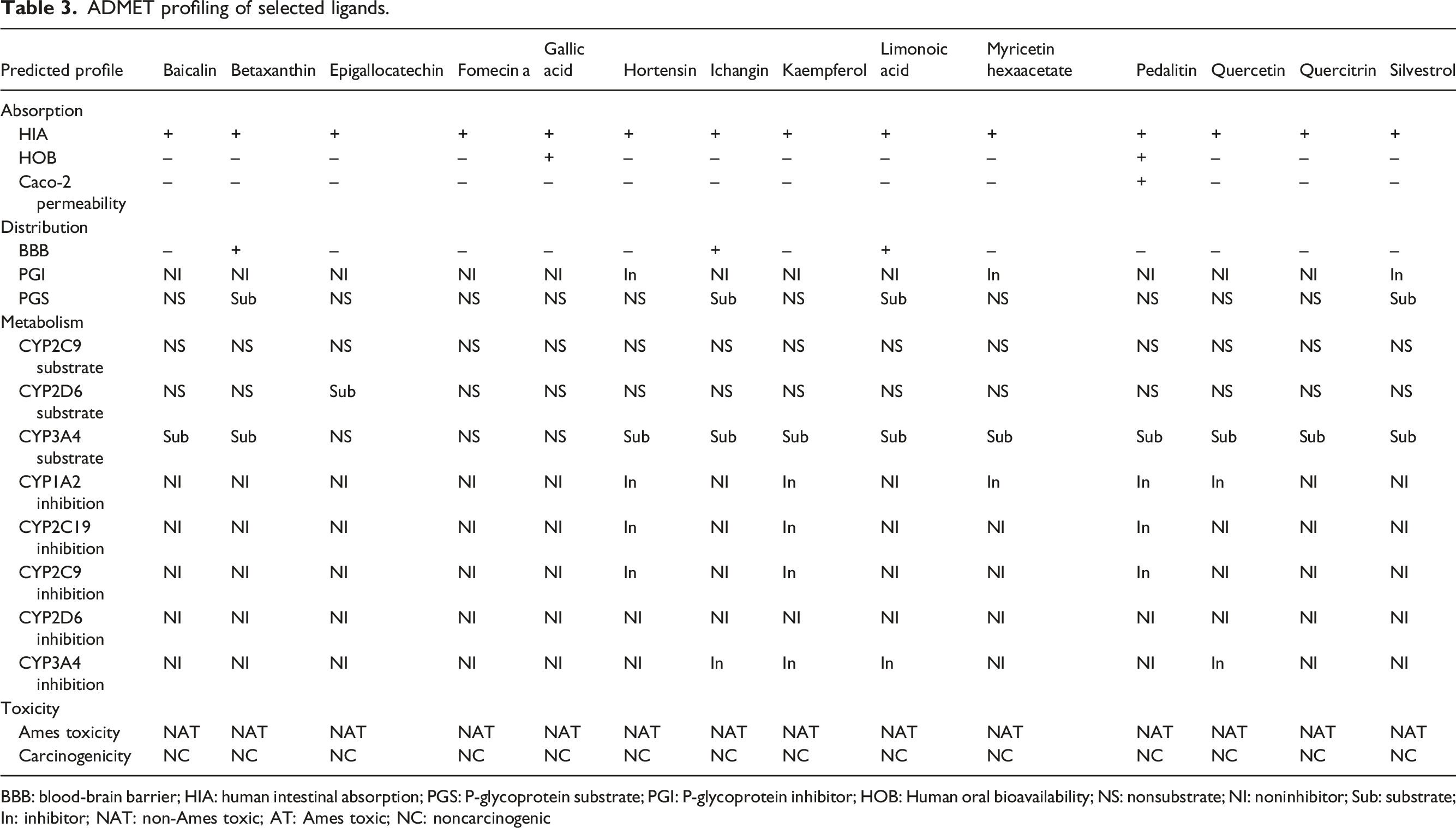
BBB: blood-brain barrier; HIA: human intestinal absorption; PGS: P-glycoprotein substrate; PGI: P-glycoprotein inhibitor; HOB: Human oral bioavailability; NS: nonsubstrate; NI: noninhibitor; Sub: substrate; In: inhibitor; NAT: non-Ames toxic; AT: Ames toxic; NC: noncarcinogenic
Molecular dynamics simulation
The evolution of RMSD values for the C-alpha atoms of ligand-bound proteins over time is shown in Figure 7. The quality of selected protein receptors (i.e., RdRp and main protease C145S mutant) were predicted using PROCHECK Ramachandran plots and shown as Figure S13 and S14. The proteins in the complex of pedalitin with SARS-CoV-2 main protease C145S mutant (MPCM-ligand) reached stability at 35 ns, according to the RMSD plot (Figure 7(a)). Following that, fluctuations in RMSD values remain within 1.0 for the duration of the simulation which is perfectly fine. Ligand fit to protein RMSD values fluctuated within 1.5 Å up to 100 ns and remained steady for the duration of the simulation. The RMSD plot of the complex pedalitin with SARS-Cov-2 RdRp (RDRP-ligand) shows that the complex reached stability at 15 ns (Figure 7(b)). Following that, fluctuations in RMSD values for protein remain within 1.5 Å during the simulation period. The RMSD values of ligands fit to proteins oscillate within 2.0 Å up to 95 ns. Then, there was a flip and an increase of 3 Å in RMSD values was observed at the end of the simulation. This indicates that during the simulation time, the ligand remains stably attached to the binding sites of both receptor proteins. Root mean square deviation (RMSD) of the C-alpha atoms of proteins and the ligand. A: MPCM-ligand complex (SARS-CoV-2 main protease C145S mutant with pedalitin), B: RDRP-ligand complex (SARS-Cov-2 RdRp with pedalitin) with time. The left Y-axis shows the variation of protein RMSD through time. The right Y-axis shows the variation of ligand RMSD through time.
The RMSF values of the proteins coupled to the ligand is shown in Figure 8. The residues with higher peaks belong to loop areas or N- and C-terminal zones, as determined by MD trajectories (Figure 9). The stability of ligand binding to the protein is shown by low RMSF values of binding site residues. Residue wise Root Mean Square Fluctuation (RMSF) of protein complexes. A: MPCM-ligand complex (SARS-CoV-2 main protease C145S mutant with pedalitin), B: RDRP-ligand complex (SARS-CoV-2 RdRp with pedalitin). Protein Secondary Structure element distribution by residue index throughout the protein structures complexed with ligand. A: MPCM-ligand complex (SARS-CoV-2 main protease C145S mutant with pedalitin), B: RDRP-ligand complex (SARS-CoV-2 RdRp with pedalitin). Red columns indicate α-helices, and blue columns indicate β-strands.

The percentages of helices and strands in MPCM-ligand complex were determined to be 16.30% and 24.37%, respectively, and the overall secondary structure elements were found to be 40.63%. Helices and strands were 35.19% and 9.65% for RDRP-ligand complex, respectively. SSE was 44.85% in total.
As seen in Figure 10, the majority of the significant ligand–protein interactions determined with MD are hydrogen bonds and hydrophobic interactions. In terms of hydrophobic interactions, His41 is the most important, whereas Thr26, His41, Cys44, and Glu166 are important in terms of H-bonds for the MPCM-ligand complex (Figure 10(a)). In terms of hydrophobic contacts, Arg349 is the most important, while Glu350, Arg457, and Asn459 are vital for H-bonds in RDRP-ligand complex (Figure 10(b)). The stacked bar charts were standardized over the course of the trajectory: for example, a value of 1.0 indicates that the specific interaction was maintained for 100% of the simulation time. Because some protein residues may make several interactions of the same subtype with the ligand, values above 1.0 are feasible. Protein-ligand contact histogram. Protein structures complexed with A: MPCM-ligand complex (SARS-CoV-2 main protease C145S mutant with pedalitin), B: RDRP-ligand complex (SARS-CoV-2 RdRp with pedalitin).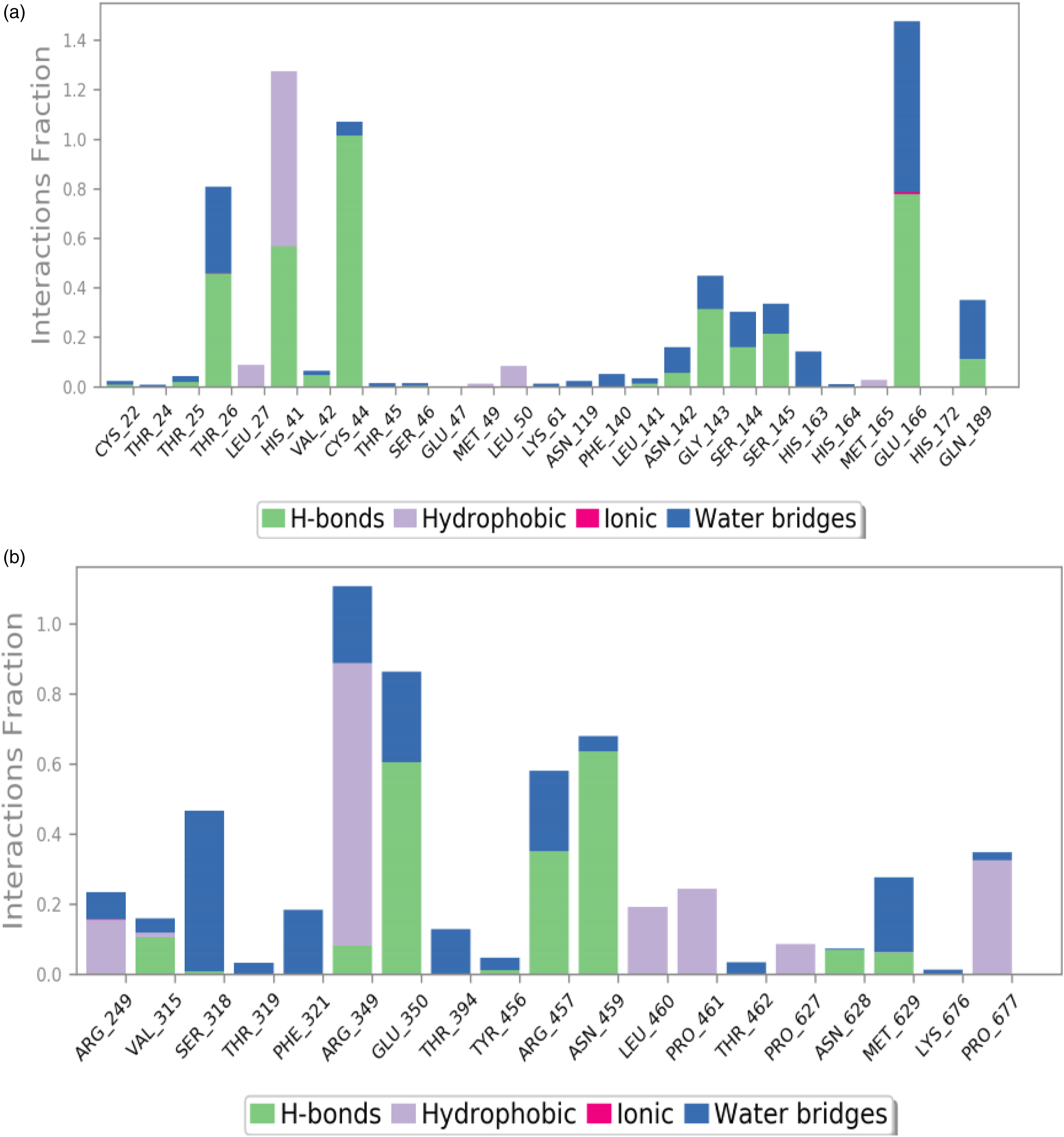
The distribution of atoms in a protein around its axis is known as the radius of gyration (Rg). Rg is the length that reflects the distance between the rotating point and the place where the energy transfer has the greatest effect. This conceptual idea also aids in the identification of diverse polymer kinds, such as proteins. The two most important markers for forecasting the structural activity of a macromolecule are the calculation of Rg and distance calculations because when a ligand/lead molecule binds to a protein, the radius of gyration changes due to a conformational shift. The folding pace of a protein is directly related to its compactness that is tracked by an advanced computer approach for determining the radius of gyration. The radius of gyration calculated for two targets bound with ligand is shown in Figure 11. Radius of Gyration calculated for two targets bound with ligand. A: MPCM-ligand complex (SARS-CoV-2 main protease C145S mutant with pedalitin), B: RDRP-ligand complex (SARS-CoV-2 RdRp with pedalitin).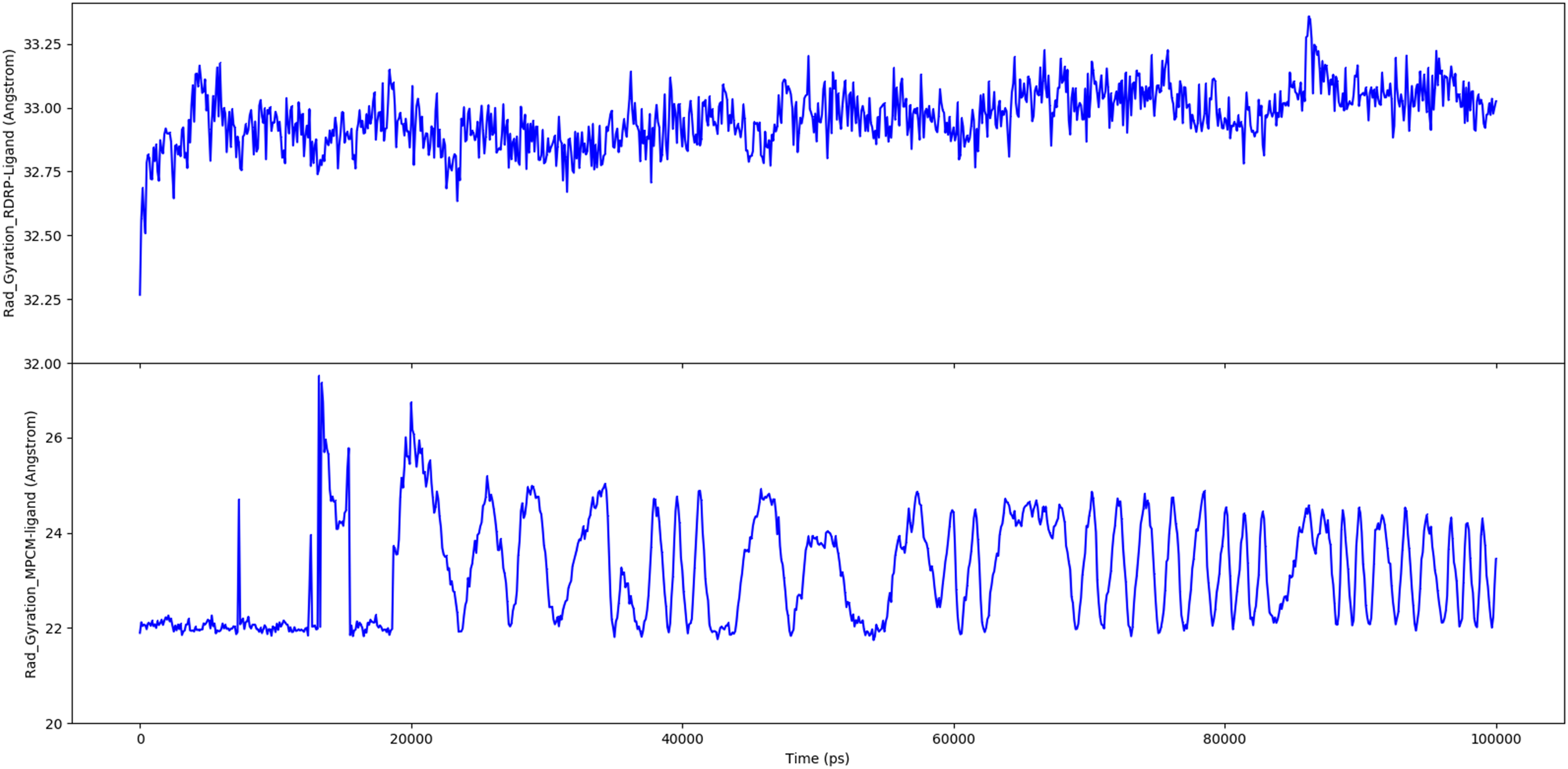
For MD Simulation, the Molecular Mechanics Generalized Born Surface Area (MM-GBSA) was determined (Figure 12). For the MPCM-ligand complex, the average dG was −65.33, the dG standard deviation was 4.63, and the dG range was −69.69 to −60.70. The average dG for RDRP-ligand complex was −67.39, with a dG standard deviation of 15.63 and a dG range of −83.03 to −51.75. It was exhibited that most of the residues of both receptors near the binding site of the ligand contributed significantly in stabilizing the ligand as can be seen by lower binding energy values. MM-GBSA calculated before and after the simulation.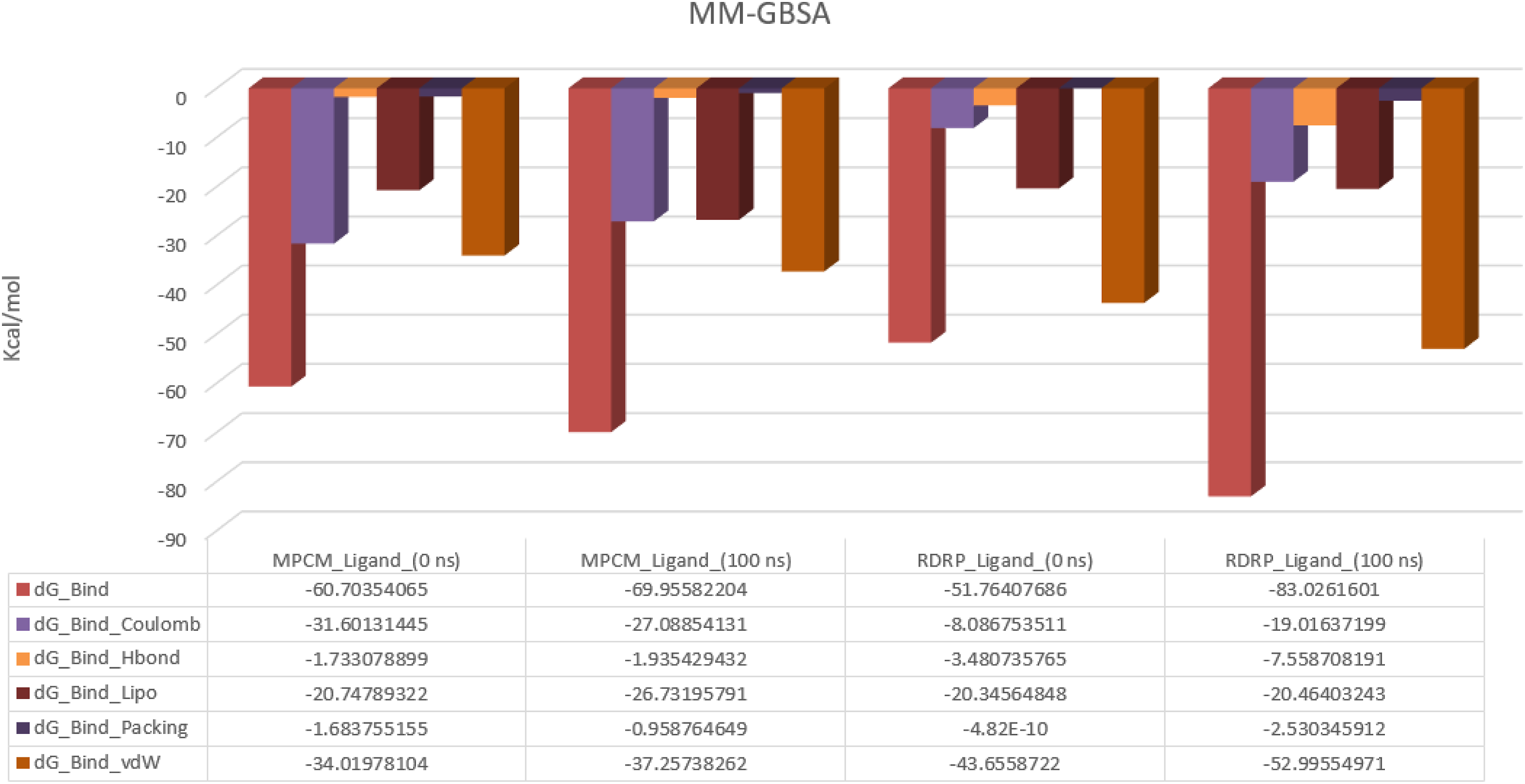
Discussion
Scientists all over the world have been working tirelessly to learn more about this new virus (SARS-CoV-2) and the pathophysiology of the disease since the current outbreak of a viral epidemic. Overall, the current understanding of the SARS-CoV-2 virus, as well as previous understanding of SARS-CoV 2003 and MERS-CoV 2012 is assisting in the development of new therapeutic agents, targets, and vaccines. 33 For decades, diverse regions and human population have used natural flora for therapeutic reasons. A wide range of phytochemicals with bioactive components have been revealed from herbal medicines which are used to live healthy and cure various diseases. 34 Herbal medicine extracts contain vitamin supplements, mineral deposits, and secondary metabolites that have fewer side effects. 35
Taking into account the nature of SARS-CoV-2 and its rising rate of infection, discovering potential therapeutic strategies and continuing to develop a vaccine is a race against time. 36 There is currently no treatment readily accessible to kill the virus or stop the infection. As a result, figuring broad-spectrum antimicrobial agents that may mitigate the effects of human coronavirus infection is incredibly hard. 37 Novel computational skills help researchers to analyze the binding interactions of various ligands prior to conducting laboratory experiments. Molecular docking and MD simulation approaches are cost-effective and cutting-edge drug discovery techniques that provide crucial data about the active site targeting and stability of potential drug candidates against specific receptor proteins. 8
In current study, we chose SARS-CoV-2 proteins that are essential for viral replication, maturation, viral entry into host cells, and some other nonstructural proteins could be served as potential targets for therapeutics. The main proteases (Mpro) are one such key protein. It is frequently discussed in literature as a potential drug development target.11,38 Only after main proteases cut the virus’s first long polypeptides into smaller pieces, they become functional. As a result, the spread of Coronavirus is heavily reliant on proteases.38,39 Noske et al. 21 used X-ray crystallography combined with biochemical data for the characterization of multiple forms of main protease of SARS-CoV-2. They revealed that the extra N-terminal residues of the main protease can cause conformational changes in the positioning of the domain three over the active site and hence obstruct the dimerization of the protein and diminish its activity. The nonstructural proteins NSP3, NSP7 with NSP8 in the conformation of a tetramer comprise two copies of each protein and NSP9 from SARS-CoV-2. These nonstructural proteins present in free or combination are key target for direct acting antivirals (DAAs) and serve as a baseline for the development of new antiviral therapeutic agents.12,38 RNA-dependent RNA polymerase is essential for the virus’s replication, transcription, and RNA processing machinery. 40 When a virus interacts with its host cell, it invades the cell and multiplies by utilizing the RNA synthesis machinery.5 RdRp encodes RdRp proteins which are essential for RNA virus replication and synthesis. Therefore, it is important to design novel drugs and nucleotide analogues have the potential to target RdRp of SARS-CoV-2 41 .
Pedalitin is a flavonoid mostly isolated from pterogyne plants previously reported as strong antiviral against Zika virus NS2B-NS3 protease 42 and hepatitis C virus. 43 Baicalin is a bioactive flavonoid glycoside that has the potential to be used in the development of a new broad-spectrum antiviral medication. 44 Betalain (betaxanthins), a yellow-orange pigment with antioxidant, antilipidemic, antibacterial, antitumor, antiviral, and anticancer properties, plays an important role in human health. 45 The epicatechin, epigallocatechin, and epicatechin gallate are main components of green tea and have several beneficial properties including antiviral activity. 46 Gallic acid is a trihydroxybenzoic acid that is classified as a phenolic acid and is found in gallnuts, sumac, witch hazel, tea leaves, oak bark, and other plants. It has been reported to be a very important common antioxidant and antiviral agent. 47 Ichangin is a terpenoid and sub-classified as limonoid previously reported as antiviral agent having potential against SARS-CoV-2 S-protein. 48
Kaempferol is a flavonoid found in the medicinal plant Kaempferol galanga L, as well as Crocus sativus, Portulaca oleracea, and other herbs. Kaempferol’s pharmacological activities such as antioxidant, antiinflammatory, anticancer, and antimicrobial activities have also been observed. 49 Citrus limonoids (limonoic acid) are tetranortriterpenoids chemicals found in citrus fruits such as oranges, lemons, grapefruits, and others. Citrus limonoids have antitumor, anti-inflammatory, antioxidative, immunomodulatory, antibacterial, antiviral, and other bioactivities. 50 Myricetin hexaacetate are in yellow needle crystals mostly isolated from the tea leaves form glycosides with glucose and galactose as myricetin-3-glucoside and myricetin-3-galactoside also have antimicrobial as well as antiviral properties. 51
Quercetin is a flavonoid that is rich in polyphenols as a potential chemo-preventive and anti-inflammatory agent. 52 Quercitrin is a flavanol group of natural compounds that has been investigated in a variety of studies for its anti-inflammation, antibacterial, antifungal, and antiviral properties. 53 Silvestrol is a flavagline-derived natural substance found in the bark of trees in the genus Aglaia, reported as broad-spectrum antiviral agent against corona and picornaviruses. 54
In the current study, among 120 compounds, the pedalitin displayed more potential with good binding interactions and S-score counter to RdRp and mutant main protease of SARS-CoV-2. Apart from pedalitin, other phytochemicals such as betaxanthin, fomecin A, gallic acid, hortensin, ichangin, kaempferol, quercetin, epigallocatechin, and limonoic acid also showed maximum potential as antiviral drug candidates counter to six receptor proteins of SARS-CoV-2. For comparison of pedalitin, two FDA approved drugs (i.e., remdesivir and nirmatrelvir) were also docked against RdRp and mutant main protease of SARS-CoV-2. The outcomes of this study strongly support the idea of pedalitin as a potential drug candidate.
Emirik 55 docked 9 drugs and 30 turmeric compounds against RdRp, Mpro, and spike glycoprotein receptor-binding domain of SARS-CoV-2. The study also included the turmeric active compounds and nine drugs against different SARS-CoV-2 proteins. Based on interactions between ligands and receptor proteins, it was concluded that turmeric compounds effectively bind with the selected proteins and form stable ligand-receptor complex which were further validated by MD simulation.
Rout et al. 56 used spice molecules as inhibitors of Mpro and RBD Spro of SARS-CoV-2. Among all the docked compounds, piperine effectively targeted the active site residues with highest binding affinity with viral proteins compared to control drugs. The free energy and stable complex formation between piperine and Mpro and RBD Spro of SARS-CoV-2 supported that piperine could be a potential inhibitor of selected viral proteins.
Singh et al. 57 used 65 active compounds from tea (Camellia sinensis) as potential drug candidates against NSP16 of SARS-CoV-2. They performed docking and simulation studies to target NSP16 protein of SARS-CoV-2. They reported six compounds including sinefungin, theaflavin, kaempferitrin, isoquercetin, epigallocatechin-3,5-di-O-gallate, epigallocatechin-3,4-di-O-gallate, and epicatechin-3,5-di-O-gallate as leading drug candidates compared to the standard inhibitor (i.e., sinefungin) against NSP16 of SARS-CoV-2.
To date, no effective medicine has been discovered against SARS-CoV-2. Antiviral drugs could target Mpro, RdRp, NSP7, NSP8, and NSP9 which are essential viral receptor proteins. The current study exhibited the highest binding affinity, drug-likeness, and stable complex formation between pedalitin and RdRp and mutant main protease of SARS-CoV-2 which proved the antiviral potential of pedalitin against both viral proteins. In spite of all these pros, the in silico studies have some limitations. The online tools/software used for bioinformatics analyses sometimes give different results for same analyses, and therefore, the researchers cannot fully rely on these predictions without further experimentations in the lab.
Conclusion
The present COVID-19 epidemic has had a significant impact on the planet. The goal of this study was to identify possible SARS-CoV-2 antagonists. Plant-derived phytochemicals were used in this investigation which have previously been found effective against several viral and bacterial proteins in the literature. Based on hydrophobic interactions, binding amino acid residues, and S-scores, three phytochemicals were selected against main proteases, RNA-dependent RNA polymerase, ADP ribose phosphatase, and nonstructural proteins NSP7, NSP8, and NSP9 from SARS-CoV-2. From drug-likeness and ADMET profiling observation, it was concluded that baicalin, betaxanthin, epigallocatechin, fomecin A, gallic acid, hortensin, ichangin, kaempferol, limonoic acid, myricetin hexaacetat, pedalitin, quercetin, quercitrin, and silvestrol exhibited strong antiviral potential against key proteins of SARS-CoV-2 which are important for viral transcription, replication, and proliferation. Moreover, the MD simulation study also exhibited thermal stability and stable binding affinity of the pedalitin with two SARS-CoV-2 receptor proteins (i.e., RdRp and main protease C145S mutant) which suggests substantial efficacy of this phytochemical against SARS-CoV-2.
Supplemental Material
Supplemental Material - Virtual screening of phytochemicals by targeting multiple proteins of severe acute respiratory syndrome coronavirus 2: Molecular docking and molecular dynamics simulation studies
Supplemental Material for Virtual screening of phytochemicals by targeting multiple proteins of severe acute respiratory syndrome coronavirus 2: molecular docking and molecular dynamics simulation studies by Muhammad Azeem, Ghulam Mustafa and Hafiza S Mahrosh in International Journal of Immunopathology and Pharmacology
Footnotes
Acknowledgments
The authors acknowledge Syed A Attique from School of Interdisciplinary Engineering & Science, National University of Sciences & Technology, Islamabad, Pakistan for assistance in molecular docking studies.
Author contributions
MA and GM conceived and designed research. MA and HSM worked on molecular docking experiments and GM worked on molecular dynamics simulation. MA and GM analyzed data. The draft manuscript was prepared by MA that was proofread by GM. HSM helped in the revision of the manuscript. All authors read and approved the final manuscript.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article
Ethics approval
The ethical approval to conduct this study is not applicable because this is an in silico study and no humans or other organisms were used in this study.
Informed consent
Not applicable as no humans or other organisms were used in this study.
Data availability
The data that support the findings of this study are available from the corresponding author, Ghulam Mustafa, upon reasonable request
Supplemental material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.