
Abstract
Introduction:
Increased oxidative stress is associated with vascular calcification in patients with chronic kidney disease (CKD). We have previously demonstrated that cellular-derived matrix vesicles (MV), but not media-derived MV, are endocytosed in the presence of phosphorus by recipient normal rat vascular smooth muscle cells (VSMC) and induce calcification through ERK1/2 and [Ca2+]i signaling. We hypothesized that these changes were mediated by increased reactive oxygen species (ROS) production.
Methods:
MV were co-cultured with recipient VSMC in the presence of high phosphorus and ROS production and cell signaling assessed.
Results:
The results demonstrated MV endocytosis led to increased ROS production in recipient VSMC with no increase in mitochondrial oxygen consumption or oxidative phosphorylation (OXPHOS), indicating the ROS was not from the mitochondria. The use of inhibitors demonstrated that endocytosis of these MV by VSMC led to a signaling cascade in the cytoplasm beginning with ERK1/2 signaling, then increased [Ca2+]i and stimulation of ROS production, mediated by nicotinamide adenine dinucleotide phosphate (NADPH) oxidase (NOX)1/4. Media-derived MV did not induce this cascade, indicating endocytosis itself was not a factor. Furthermore, inhibition of either ERK1/2 activation or [Ca2+]i reduced vascular calcification.
Conclusion:
We conclude that endocytosis of pro-mineralizing MV can induce a series of signaling events in normal VSMC that culminate in generation of ROS via activation of NOX1/4. Understanding these pathways will allow the development of future targeted therapeutics.
Keywords
Introduction
Vascular calcification in coronary and peripheral arteries affects most patients with advanced kidney disease and can manifest as ischemia leading to myocardial infarction or left ventricular hypertrophy due to increased afterload from aorta calcification, and sudden cardiac death due to calcification of arterioles in the myocardium.1,2 There are many clinical risk factors associated with calcification including older age, hyperphosphatemia, hypertension, calcium-containing phosphate binders, and oxidative stress.2,3 The strongest risk factor for progressive calcification is existing arterial calcification, 4 suggesting that there is a mechanism by which calcification can expand, or propagate, within a vessel. We have developed an in vitro model of vascular smooth muscle cells (VSMC) calcification/propagation where matrix vesicles (MV) derived from calcifying VSMC from a rat model of chronic kidney disease (CKD) are co-cultured with VSMC from normal littermates to induce the propagation of calcification of the normal cells. 5 The endocytosis of cellular-derived MV by normal VSMC accelerates the time course of calcification from 10 to 14 days to 4 to 7 days, whereas media-derived MV did not accelerate calcification. 5 Furthermore, the cellular-derived MV had increased expression of CD63 and annexin II, V, and VI, whereas media-derived MV had greater amounts of fetuin (natural calcification inhibitor), CD8, and CD9. Neither source of MV expressed alkaline phosphatase. 5 These differences allow us to compare the role of these different types of MV, both of which originate from VSMC undergoing calcification.
Our previous studies demonstrated that both cellular and media-derived MV can be endocytosed by recipient normal VSMC. However, only cellular MV induced an acute increase in [Ca2+]i and activation of the ERK1/2 signaling pathway in recipient VSMC. We also found NOX1 expression was increased in recipient VSMC, and that blockade of NOX1/4 activity with GKT137831 decreased calcification. 5 However, the sequence of events and interplay of these three signaling pathways is not known. NOX is involved in the regulation of VSMC phenotype,6–8 and conversion from a contractile to synthetic state of VSMC is a critical step in the pathogenesis of vascular calcification. 9 The function of the NOX family is to catalyze the reduction of O2 to produce superoxide (O2–), which in turn generates hydrogen peroxide, leading to generation of reactive oxygen species (ROS). 10 Mitochondria are a target of NOX-induced ROS and serve as a regulator of overall cellular redox status through production of antioxidants, but mitochondria also produce ROS that can activate NOX, leading to complex cellular regulation.10,11 Thus, although ROS is involved in the pathogenesis of vascular calcification, the regulation remains poorly defined. We hypothesized that MV-induced generation of ROS from NOX may be the initial response in the recipient VSMC leading to downstream cell signaling and enhanced calcification.
Materials and methods
Animal models and cell culture
Primary rat VSMC were isolated from a model of chronic kidney disease-mineral bone disorder (CKD-MBD), the Cy/+ rat. This model spontaneously develops all three manifestations of CKD-MBD: biochemical abnormalities, extraskeletal calcification, and abnormal bone.12–18 All animal procedures were approved by the Indiana University School of Medicine’s Institutional Animal Care and Use Committee prior to initiating the study. VSMC were isolated from the descending thoracic aorta in CKD rats by the explant method. Normal VSMC were similarly derived from normal littermates (Cy–/–). To induce calcification, rat VSMC were treated with 5 mM β-glycerophosphate, 1 U/mL fetal alkaline phosphatase, and 15% fetal bovine serum (calcification media).
Matrix vessels (MV) isolation
Cellular-derived MV were isolated using our previously published method. 5 Briefly, cellular MV were isolated by collagenase digestion followed by sequential centrifugation. For some experiments, media MV were isolated from the cell media by sequential centrifugation and used as a control as these do not induce recipient VSMC calcification. 5 Each set of MV is isolated from VSMC pooled from three CKD rats and stored in Tris buffered saline (pH 7.5 with 0.25 M sucrose). Quantitation of MV was determined by protein concentration.
MV-VSMC co-culture
The normal VSMC were incubated in calcification media at the time of the addition of MV, and then incubated for different durations with or without 10 μg of cellular MV (known to induce calcification) or media MV (known to not induce calcification). MV were isolated from CKD rat VSMC in the presence of calcification media containing β-glycerophosphate. 5 Protein was isolated to identify changes in autophagy or activation of ERK1/2 from the recipient cells. In some experiments, MV-VSMC co-cultures were treated with or without an inhibitor of inositol 1,4,5-trisphosphate (IP3)-receptor 2-APB (Calbiochem, Darmstadt, Germany), ERK1/2 inhibitor U0126 (Cell Signaling Technology, Danvers, MA, USA) or NOX1/4 inhibitor GKT137831 (Biovision, Milpitas, CA, USA). Calcification was quantified by the o-cresolphthalein complexone method.5,19,20
Measurements of ROS generation in recipient VSMC
Cellular or media MV (10 μg) were incubated with VSMC for 24 hours in calcification media and the ROS generation in VSMC was assessed using CellROX Oxidative Stress Reagents (Molecular Probes, Eugene, OR, USA) by confocal microscopy. Confocal images of 8–10 randomly chosen fields per sample (each dish) were acquired with an Olympus FV1000 MPE confocal/2-photon system (Olympus, Center Valley, PA, USA) in the Indiana Center for Biologic Microscopy, Indianapolis, IN, using an Olympus UPlanSApo 60×/1.2 objective lens. Images were analyzed and quantified using Metamorph software, v.7.7 (Molecular Devices, San Jose, CA, USA). Initial studies demonstrated a 30% increase in ROS production in normal VSMC incubated just with beta-glycerophosphate-containing (calcification) media, even in the absence of MV. Therefore, we only assessed the effect of MV on induction of ROS in calcification media. Given the differences in overall fluorescence intensity between experiments, each experiment assessed 8 to 10 fields in each dish/sample (control – no MV, + cellular MV, + media MV) for confocal fluorescence intensity. The 8 to 10 fields assessed in each dish were averaged, and then represented as the % control dish (no MV) for each experiment. In additional experiments, cellular MV (10 μg) were incubated with VSMC for 24 hours in the presence of mitochondrial-targeted antioxidant (400 µM MitoTEMPO; Sigma-Aldrich, St Louis, MO, USA); inhibitor of the ER chaperones protein disulfide isomerase (PDI) bacitracin (500 µM); NOX1/4 inhibitor GKT137831 (4 µM) or IP3 receptor inhibitor (2-APB, 10 µM); and the ROS generation in VSMC was assessed using CellROX Oxidative Stress Reagents (Molecular Probes) by confocal microscopy.
Direct measurement of mitochondrial generated ROS and mitochondrial function
To determine if MV-induced cellular ROS in VSMC is generated from the mitochondria, cellular MV (10 μg) were first incubated with VSMC for 1 or 24 hours and the mitochondria ROS production in VSMC examined using MitoROS 580 (Cayman Chemical Company, Ann Arbor, MI, USA) according to the manufacturer’s instructions. To confirm that VSMC will respond and generate mitochondrial ROS, VSMC were treated with the positive control antimycin A (AMA; 50 µM and 250 µM) for 24 hours. The mitochondrial ROS production was examined at 540/590 nm Ex/Em using CLARIOstar high performance microplate reader (BMG LABTECH Inc., Cary, NC, USA).
To determine if MV affect mitochondrial function, VSMC were counted using Trypan blue with hemocytometer. Cells were seeded (1 × 104/well) in an XF24 cell culture microplate and incubated with or without cellular MV (10 μg) for 24 hours and mitochondrial function in recipient VSMC examined using the XF24 Extracellular Flux Analyzer from Seahorse Bioscience (North Billerica, MA, USA). 21 After 24 hours of MV-VSMC co-culture, the media was replaced with prewarmed XF24 assay media for 1 hour; this unbuffered media consisted of DMEM (Sigma), 1 mM Glutamax-1 (Invitrogen), 2 mM pyruvate, 141 mM NaCl, and 25 mM glucose. Three basal oxygen consumption rates (OCR) were measured to obtain a baseline OCR, followed by the sequential injection. ATP production was calculated in response to oligomycin (1 μg/mL). Maximum and reserve respiration capacity was assessed with carbonyl cyanide-4-(trifluoromethoxy)phenylhydrazone (FCCP, 1 μM) and AMA (10 μM) after each intervention. In a parallel experiment, we plated an equal number of VSMC into wells and demonstrated the addition of MV or no MV had no effect on total protein after 24 hours (VSMC with no MV, total protein is 25.14 ± 1.71 µg; VSMC + MV, total protein is 25.23 ± 2.42 of µg) and therefore no protein normalization was done.
Assessment of mitochondrial antioxidants
Nuclear and cytosolic protein were isolated using Cayman’s Nuclear Extraction kit (Cayman Chemical Company) from recipient VSMC. Nuclear factor erythroid 2-related factor 2 (Nrf2) was measured in the nuclear fraction, and the major regulator of Nrf2, Keap1 (Kelch-like ECH-associated protein I), measured in the cytosolic fraction with antibody against Nrf2 or Keap 1, respectively (1:1000; Santa Cruz Biotechnology, Santa Cruz, CA, USA), by Western blot. Primary antibody incubation was at 4°C overnight and was followed by incubating with peroxidase conjugated secondary antibody (1:5000 dilution) and immunodetection with the Enhanced Chemiluminescence Prime Western Blot Detection Reagent (Amersham, Piscataway, NJ, USA). Band intensity was analyzed by the ChemiDoc MP Imaging System (Imaging Lab 4.0; Bio-Rad, Richmond, CA, USA) utilizing a proprietary polyacrylamide gel chemistry to make proteins fluorescent in the gel allowing visualization and quantitation of total protein for normalization.
ERK1/2 activation in VSMC by Western blot
The activation of ERK1/2 was assessed using Western blot as previously published. 5 Briefly, 20 µg of protein was loaded on 10% SDS-PAGE and the blots incubated with antibody against Phospho-ERK1/2 (1:500; Cell Signaling Technology) overnight at 4°C followed by incubating with peroxidase conjugated secondary antibody (1:5000 dilution), and immunodetection with the Enhanced Chemiluminescence Prime Western Blot Detection Reagent (Amersham). For loading control, western blot was performed using antibodies against total ERK1/2 (1:1000; Cell Signaling Technology). The band intensity was analyzed by the ChemiDoc MP Imaging System (Imaging Lab 4.0; Bio-Rad) and ERK1/2 activation was quantified by normalizing phosphorylated ERK1/2 to total ERK1/2.
RNA isolation and real-time PCR for miRNA expression
Total RNA from MV-VSMC was isolated using a miRNeasy Mini Kit (Qiagen, Germantown, MD, USA) according to the manufacturer’s instructions. MicroRNA expression was determined by real-time polymerase chain reaction (PCR) using TaqMan miRNA Assays (Applied Biosystems, Foster City, CA, USA) as we have previously published. 22 Target-specific PCR primers (miR-21, miR-29b, miR-155, miR-210, and miR-424) were obtained from Applied Biosystems. Real-time PCR amplification was performed using TaqMan miRNA Assays (TaqMan MGP probes, FAM dye-labeled) using ViiA7 Real-Time PCR systems (Applied Biosystems). The cycle number at which the amplification plot crosses the threshold was calculated (CT), and the ∆∆CT method are used to analyze the relative changes in gene expression and normalized by U6, a nonhuman ubiquitous miRNA.
Analysis of autophagy by Western blot
Proteins were isolated from MV-VSMC co-culture at days 1, 3, and 7 and Western blot performed. The blots were incubated with antibodies against autophagy markers LC3B and Atg5 (1:750 and 1:1000, respectively; Cell Signaling Technology) followed by incubation with peroxidase-conjugated secondary antibody (1:5000 dilution) and immunodetection with an enhanced chemiluminescence kit. The band intensity was analyzed by the ChemiDoc MP Imaging System (Bio-Rad) and normalized to total protein as above.
Statistics
Statistical analysis was conducted by one-way ANOVA and within-group comparisons using Dunnett’s post hoc analysis. Two-group comparison was performed using unpaired t-test. The results are expressed as means ± SD, with p < 0.05 considered significant (GraphPad Prizm Software, La Jolla, CA, USA).
Results
MV increase the production of cellular ROS in recipient VSMC
To determine if MV induce ROS production in the recipient VSMC, cellular or media MV were co-cultured with normal VSMC in the presence of calcification media (5 mM β-glycerolphosphate and normal calcium) for 24 hours and cellular oxidative stress determined. Figure 1A demonstrates that cellular MV increased ROS production by fourfold at 24 hours compared to VSMC incubated with MV derived from the media (control for endocytosis), or no MV. Given the absence of ROS induced by endocytosis of media-derived MV in recipient VSMC, and our previous work that media-derived MV did not accelerate calcification, 5 all further studies were only done with the cellular-derived MV, hereafter simply termed MV.
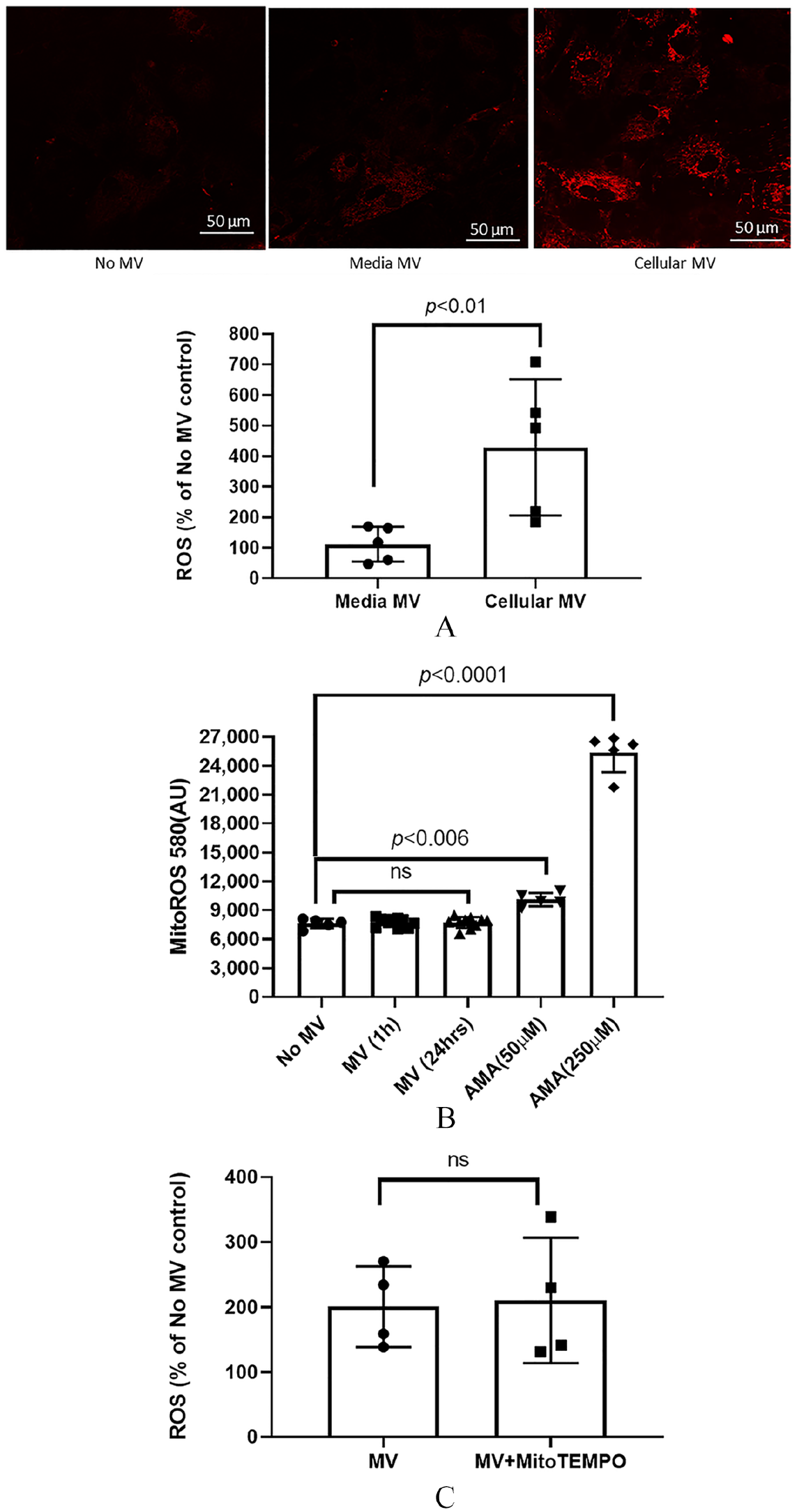
Cellular-derived MV increased the production of cellular ROS but had no effect on mitochondria ROS production or oxidative phosphorylation in recipient VSMC. Cellular (= calcifying, 10 µg) or media-derived (noncalcifying or control, 10 µg) MV were co-cultured with normal VSMC in the presence of high phosphorus (calcification) media for 24 hours and cellular ROS generation assessed using CellROX Oxidative Stress Reagents by confocal microscopy and quantified.
Cellular source of ROS
ROS can be generated from the mitochondria or cytosolic sources. To determine if the source of the increased cellular ROS was from the mitochondria, we examined mitochondrial superoxide production in VSMC using MitoROS 580. The addition of MV to normal VSMC for 1 or 24 hours had no effect on mitochondrial ROS (MitoROS 580) production in recipient VSMC, whereas the positive control AMA at 24 hours (50 µM and 250 µM) significantly increased mitochondrial ROS production in VSMC (Figure 1B). Furthermore, in the presence of mitochondrial-targeted antioxidant (MitoTEMPO), there is no change in MV-stimulated cellular ROS production in the recipient VSMC (Figure 1C). We then incubated MV with VSMC for 24 hours and examined mitochondrial respiration in VSMC using the Seahorse Extracellular Flux Analyzer. The addition of MV had no effect on basal OCR, maximal OCR, ATP production or respiratory capacity in recipient VSMC (Figure 2). These results demonstrate that the increased ROS in recipient VSMC is not generated from the mitochondria.
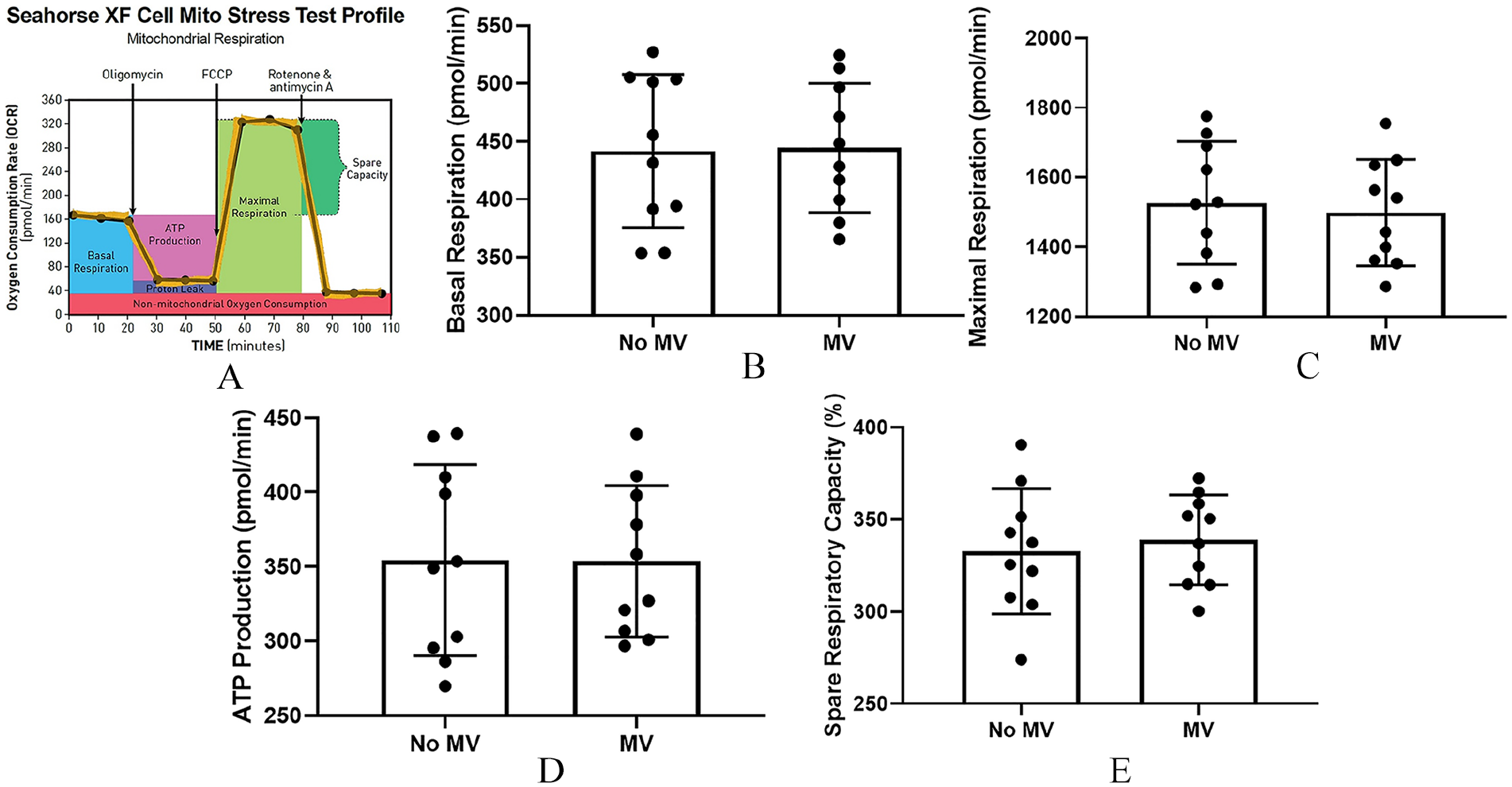
MV had no effect on mitochondrial function in recipient VSMC. Cellular MV (10 μg) were co-cultured with normal VSMC in the presence of high phosphorus (calcification) media for 24 hours and mitochondrial function in recipient VSMC examined using the Seahorse Bioscience XF24 Extracellular Flux Analyzer.
To determine if antioxidant dysregulation is a factor in the increased ROS in recipient VSMC, we assessed the master antioxidant transcription factors Nrf2/Keap1 in the cytoplasm, which is known to translocate Nrf2 to the nucleus to regulate antioxidants. The addition of MV to VSMC did not alter this pathway (online Supplemental Figure 1), indicating the increased ROS was not due to mitochondrial down-regulation of antioxidants. The increased ROS may also be from the endoplasmic reticulum (ER); however, blockade of PDI with bacitracin to inhibit ER function did not alter MV-induced ROS production in recipient VSMC (Figure 3A). However, blocking NOX1/4 activity with GKT137831 decreased MV-induced ROS production in recipient VSMC (Figure 3B). These data suggest that the increased ROS originates from NOX activation in recipient VSMC that is not from the mitochondria or due to a reduction in Nrf2 regulated antioxidants.
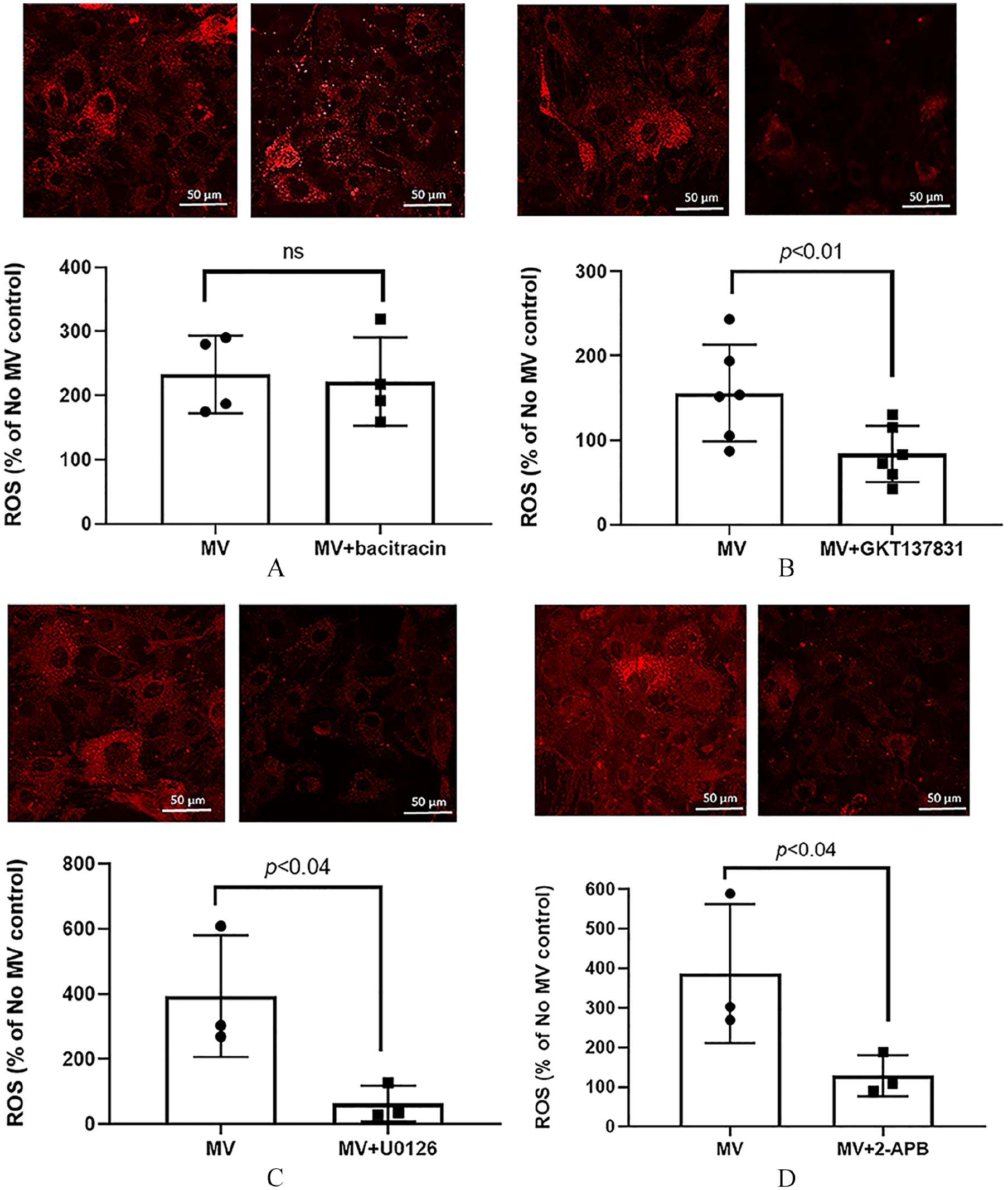
Regulation of ROS production. The effect of inhibition of the ER chaperones PDI, NOX1/4 activity, ERK1/2 activity or intracellular calcium release on MV-induced ROS production in recipient VSMC. MV (10 μg) were added to normal VSMC in the presence or absence of inhibitors for 24 hours and ROS generation assessed with CellROX Oxidative Stress Reagents by confocal microscopy.
Interplay of cytoplasmic cell signaling pathway
ROS can be an upstream response to increased [Ca2+]i and ERK1/2 signaling, or a downstream response to these same pathways. To determine if increased ROS is mediated by the ERK1/2 signaling pathway, cellular MV were added to normal VSMC with calcification media in the presence or absence of ERK1/2 inhibitor U0126 for 24 hours and cellular ROS production examined. Inhibition of ERK1/2 activity significantly decreased cellular ROS production in recipient VSMC at 24 hours (Figure 3C). MV were then added to normal VSMC with calcification media for 24 hours in the presence or absence of an inhibitor of IP3 receptors (2-APB), a second messenger in signal transduction of intracellular calcium. The results demonstrated that blocking IP3-mediated [Ca2+]i release also prevented MV-increased cellular ROS in recipient VSMC (Figure 3D).
To assess the importance of ERK1/2 signaling or intracellular calcium release in MV-induced calcification in recipient VSMC, MV were added to normal VSMC with calcification media in the presence or absence of the ERK1/2 inhibitor U0126 or the IP3 receptor inhibitor (2-APB) for 4 days and calcification determined. Both inhibitors decreased MV-induced calcification in recipient VSMC (Figures 4A and 4B). To identify the sequence of events, MV were added to normal VSMC in the presence or absence of 2-APB and activation of ERK1/2 determined by Western blot. The results demonstrated that blocking IP3-mediated [Ca2+]i release had no effect on MV-induced ERK1/2 activation in recipient VSMC (Figure 5A). Of note, U0126 can also block ERK5, but we did not find increased activation of ERK5 with the addition of MV in recipient VSMC (online Supplemental Figure 2). Furthermore, blocking NOX1/4 activity with GKT137831 also had no effect on MV-stimulated ERK1/2 activation in recipient VSMC (Figure 5B). In contrast, we have previously demonstrated that inhibition of ERK1/2 decreased MV-induced intracellular calcium release in recipient VSMC. 5 This indicates that MV-induced activation of ERK1/2 is likely the initial signaling event in the generation of ROS.
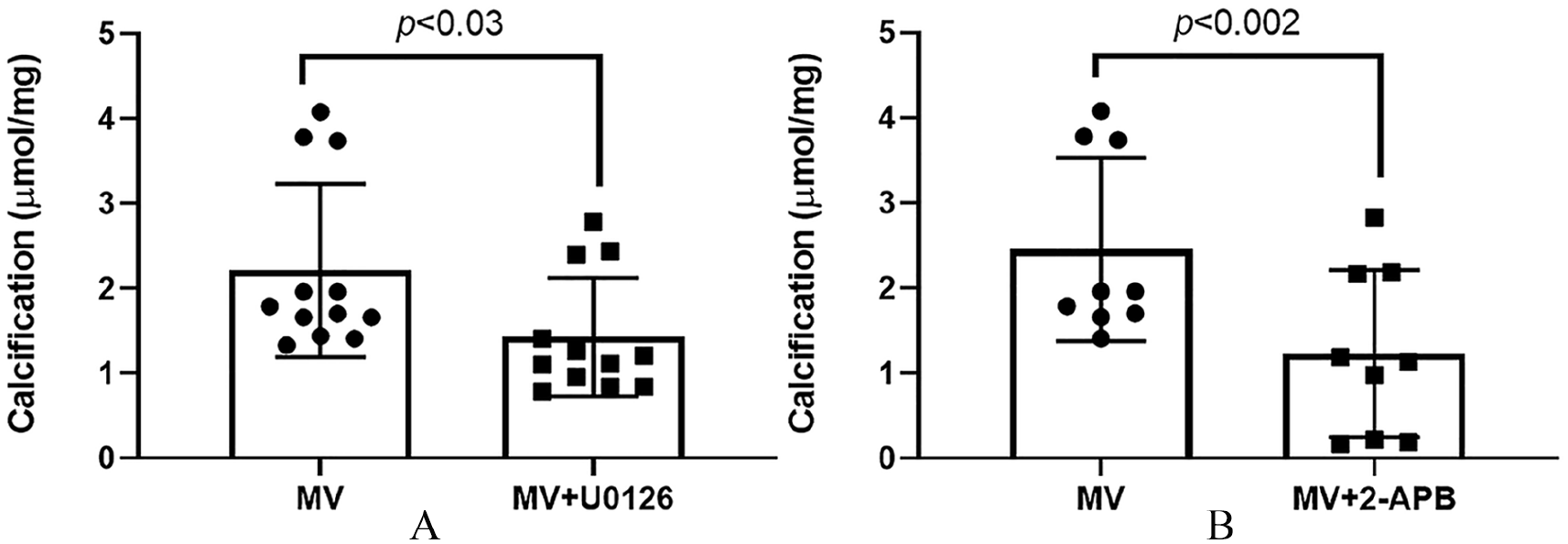
The roles of ERK1/2 and intracellular calcium on MV-induced calcification in recipient VSMC. MV (10 µg) were added to normal VSMC with calcification media in the presence or absence of ERK1/2 inhibitor U0126 or IP3 receptor inhibitor (2-APB) for 4 days and calcification determined. The results demonstrated that
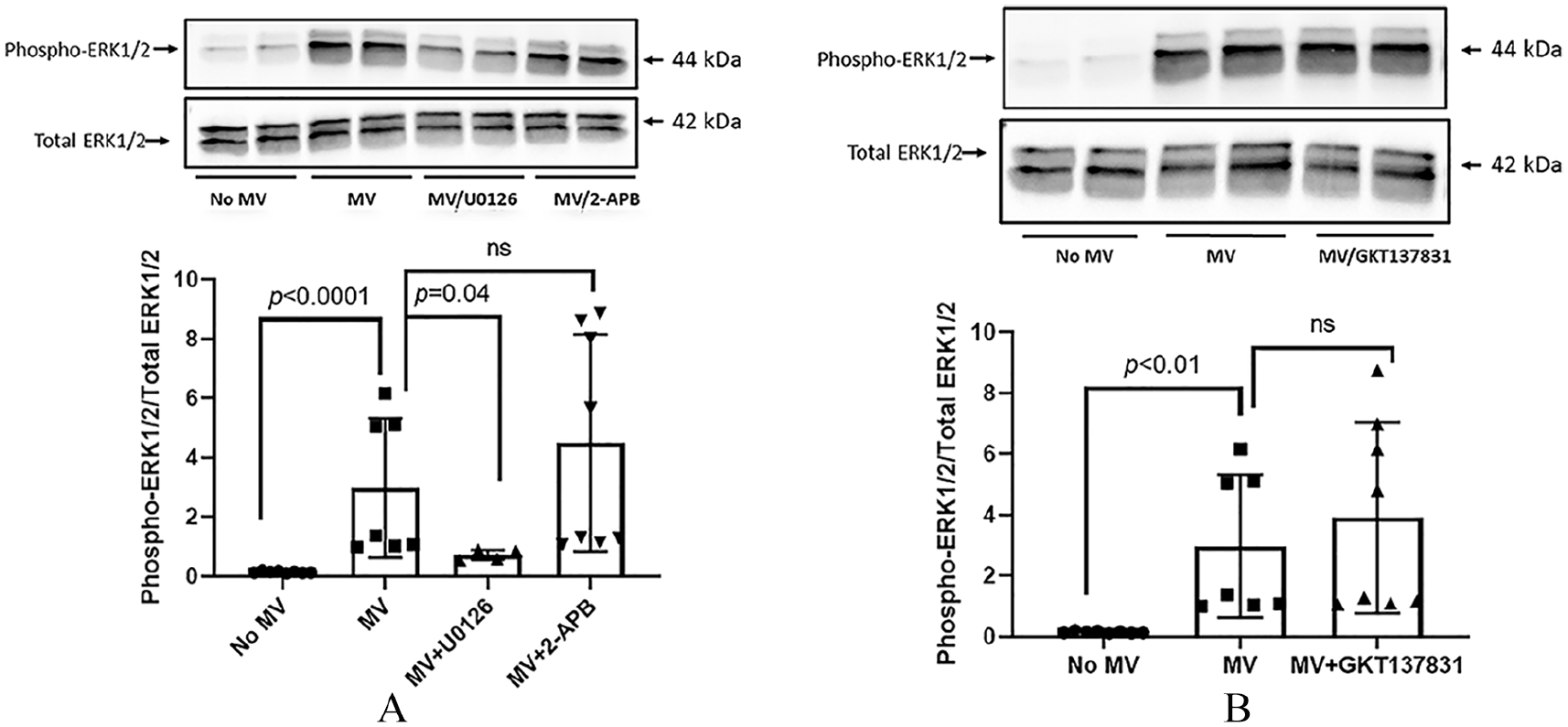
Inhibition of intracellular calcium release or NOX1/4 activity had no effect on MV-induced activation of ERK1/2 in recipient VSMC. To further elucidate MV-induced signaling cascade, MV (10 µg) were added to normal VSMC with calcification media in the presence or absence of IP3 receptor inhibitor (2-APB, 10 µM), ERK1/2 inhibitor U0126 (10 µM), or NOX1/4 inhibitor (GKT137831, 4 μM) and activation of ERK1/2 determined by Western blot.
Alternative regulators of recipient VSMC ROS production and calcification
The endocytosis of MV by recipient VSMC may also activate other pathways that have been shown to be involved in ROS generation and/or vascular calcification. Multiple studies have indicated that dysregulation of specific miRNAs can lead to oxidative stress-ROS production, inflammation, and atherosclerosis.23,24 We therefore examined the expression of these known ROS-regulating miRNA in recipient VSMC at 24 hours and found that MV did not alter the miRNA expression (online Supplemental Figure 3). We also examined the roles of autophagy, as oxidative stress can induce autophagy25,26 as can high phosphorus media. 27 We cultured VSMC with control media (no additional phosphorus), calcification media (high phosphorus) alone, or calcification media with added MV for up to 7 days and assessed expression of autophagy markers Atg5 and LC3B-II/LC3B-I at days 1, 3, and 7 during calcification, using rapamycin as a positive control. However, neither high phosphorus media alone, nor the addition of MV in calcification media induced autophagy at any time point (online Supplemental Figure 4). These results suggest that MV endocytosis-induced ROS is not due to autophagy or upregulation of miRNA.
Discussion
Vascular calcification remains a significant cause of morbidity and mortality in CKD, diabetes, and aging. 3 In bone, MV released from cartilage or bone cells induces mineralization on a matrix of collagen, and ROS generation is involved in phosphorus-induced mineralization in cultured osteoblast cell lines. 28 We have similarly identified that MV from VSMC can mineralize collagen in a calcium and phosphorus dependent manner, even in the absence of cells. 20 We hypothesized that the converse can also occur: propagation, or expansion, of arterial medial mineralization from calcified VSMC to normal, healthy VSMC may be from cell signaling events induced by the endocytosis of MV by normal VSMC. We have previously shown that this form of cell–cell communication induces calcification in the recipient cells through ERK1/2 and [Ca+2]i signaling, and blockade of NOX1/4 could inhibit calcification. 5 The current study expands this work and determines the sequence of events. We identified that increased ROS, as a result of ERK1/2-induced changes in [Ca2+]i, is the likely mechanism. Importantly, these cell signaling changes were not just from the endocytosis, as media-derived MV did not induce this cascade. Further, the source of the increased ROS is not from the mitochondria as assessed by direct respirometry and OXPHOS, suggesting that endocytosis of these MV sets off a detrimental signaling cascade in the cytoplasm, beginning with ERK1/2 signaling, then release of [Ca2+]i and stimulation of ROS production mediated by NOX1/4 (Figure 6).
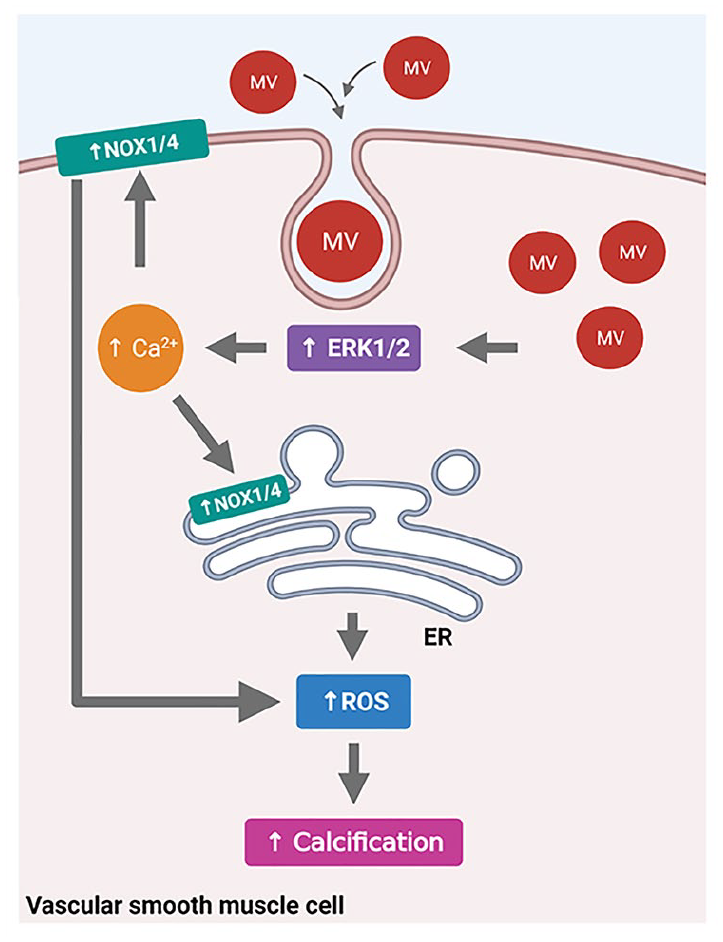
Overview of MV-induced cell signaling, ROS production, and calcification in recipient VSMC. Cellular-derived MV can be endocytosed into normal VSMC, which induces the activation of ERK1/2 and increased release of [Ca2+]i in VSMC. This activates NOX1 and/or NOX4 at either the endoplasmic reticulum or at the cell membrane/focal adhesions, resulting in increased cellular ROS production and enhanced calcification of VSMC.
Cellular ROS generation is complex and difficult to quantify due to the presence of antioxidants. Our previous work found MV-induced increased expression of SOD-2 only at 3 days in recipient VSMC whereas the NOX1 production was observed as early as 1 day, 5 and in the current study we did not find alteration in the antioxidant master regulator system of Nrf2/Keap1. These data suggest that the generation of ROS was an early event and not a result of deficient antioxidants. Further, we identified that mitochondria were not the source of ROS, but rather that the endocytosis of the MV generated increased ROS.
Endocytosis is a highly conserved mechanism for regulation of receptor-mediated signaling from the plasma membrane, and is known to be associated with MAP kinase signaling. 29 In osteoblasts, endocytic cell trafficking regulates both osteoclast and osteoblast function, the degradation of mineralized matrix, and synthesis of matrix vesicles for mineralization. 30 In VSMC, we have previously shown the importance of endocytosis of MV to enhance mineralization in vitro, a process that was decreased with a NOX1/4 inhibitor. 5 This is consistent with another study that demonstrated the uptake of the TNF-alpha-ligand/receptor complex in VSMC generates ROS that can be inhibited by blockade of either NOX1 or dynamin (blocker of endocytosis). 31
Furmanik recently demonstrated that calcium loaded MV endocytosis of human VSMC led to NOX5-induced ROS generation and phenotypic switch to osteochondrocytic-like cells. 7 NOX5 is only found in humans, and, along with intestinal NOX (Duox), is regulated by calcium. 32 In the present study, we found that MV (that are filled with calcium and phosphorus) 20 upregulate NOX1-induced ROS. Interestingly, calcium load and hypercalcemia are clinical risk factors for vascular calcification. 33 The incubation of VSMC in vitro with calcium increases calcification 34 whereas we have shown that blockade of extracellular calcium entry by verapamil or annexin inhibitors decreases calcification.19,20 Taken together, these results suggest that the induction of VSMC calcification by extracellular calcium may be due to the following sequence of events: (1) increased extracellular calcium and influx of calcium into VSMC; (2) upregulation of NOX5-induced ROS (in humans) or MV-induced NOX1-induced ROS (in rats); and (3) ERK1/2 signaling with a rise in intracellular calcium. Interestingly, in the current study, we found that MV derived from the media did not induce ROS production in recipient VSMC and thus we used these as controls for endocytosis, as we have shown in cell entry/uptake by confocal microscopy (unpublished observation). Further, these vesicles contain dramatically more fetuin-A than cellular-derived MV, 5 and that fetuin-A was endocytosed via annexin calcium channels in VSMC only in the presence of extracellular calcium. 35 It is therefore plausible that the increased uptake of fetuin-A into cells occurs with elevated extracellular calcium to increase fetuin-A loading into released MV to inhibit calcification. Alternatively, the elevated fetuin-A may alter annexin channels known to modulate calcium influx. This suggests an active regulation through calcium of both the content and the endocytosis of MV by extracellular and intracellular calcium. More studies are needed to clarify this regulation and the role of concomitant increases in phosphorus.
Study limitations
There are several limitations to the current study. First, NOX can induce multiple cellular responses, including autophagy.26,27 We found no evidence of autophagy in the current study, and in contrast to Dai et al., did not find that phosphorus alone (no vesicles) induced autophagy. However, we did not fully investigate mitophagy and other similar pathways. 27 Second, we did not further characterize the specific content of MV that led to the increased ROS in the present study, although we have previously characterized the MV content in these vesicles.5,20 We also measured known miRNA that induce ROS in VSMC and found that these were not increased in the recipient VSMC, although it is possible there are other miRNA involved. We have previously also shown that MV contain miRNA, 36 and, in cancer cells, transfer of miRNA from exosomes into recipient cells has been demonstrated. 37 However, we have been unable to show that miRNA directly transfers from inside the MV into VSMC using fluorescently labeled exosome RNA with Exo-Glow Tracking Labels (System Biosciences, Palo Alto, CA, USA) (unpublished data) and thus we did not test if changing the miRNA in the vesicles altered the ROS production. Importantly, vesicles are certainly not all equivalent,38,39 and the primary role of matrix vesicles characterized by high intracellular calcium and phosphate concentrations is mineralization.
Conclusion
In conclusion, normal VSMC can endocytose pro-mineralizing MV derived from calcifying VSMC, which induce a series of signaling events: ERK1/2 phosphorylation resulting in increased intracellular calcium and generation of ROS by activation of NOX1/4 in the cytoplasm. The hypothetical mechanistic pathway of MV-induced ROS production and calcification in recipient VSMC is illustrated in Figure 6. We did not confirm the cytoplasmic location of the NOX1/4, but other studies have found that NOX1 can also be localized with the cell membrane and NOX4 can be localized in focal adhesions, 40 and a more recent paper found that NOX1 was only in the cell membrane and NOX4 only in the endoplasmic reticulum, 41 and thus the figure shows both NOX1/4 in the ER and NOX1/4 at or near the cell membrane. Understanding these pathways will allow the future development of targeted therapeutics.
Supplemental Material
sj-tif-1-vmj-10.1177_1358863X211024721 – Supplemental material for Regulation of reactive oxygen species in the pathogenesis of matrix vesicles induced calcification of recipient vascular smooth muscle cells
Supplemental material, sj-tif-1-vmj-10.1177_1358863X211024721 for Regulation of reactive oxygen species in the pathogenesis of matrix vesicles induced calcification of recipient vascular smooth muscle cells by Neal X Chen, Kalisha D O’Neill, James M Dominguez and Sharon M Moe in Vascular Medicine
Supplemental Material
sj-tif-2-vmj-10.1177_1358863X211024721 – Supplemental material for Regulation of reactive oxygen species in the pathogenesis of matrix vesicles induced calcification of recipient vascular smooth muscle cells
Supplemental material, sj-tif-2-vmj-10.1177_1358863X211024721 for Regulation of reactive oxygen species in the pathogenesis of matrix vesicles induced calcification of recipient vascular smooth muscle cells by Neal X Chen, Kalisha D O’Neill, James M Dominguez and Sharon M Moe in Vascular Medicine
Supplemental Material
sj-tif-3-vmj-10.1177_1358863X211024721 – Supplemental material for Regulation of reactive oxygen species in the pathogenesis of matrix vesicles induced calcification of recipient vascular smooth muscle cells
Supplemental material, sj-tif-3-vmj-10.1177_1358863X211024721 for Regulation of reactive oxygen species in the pathogenesis of matrix vesicles induced calcification of recipient vascular smooth muscle cells by Neal X Chen, Kalisha D O’Neill, James M Dominguez and Sharon M Moe in Vascular Medicine
Supplemental Material
sj-tif-4-vmj-10.1177_1358863X211024721 – Supplemental material for Regulation of reactive oxygen species in the pathogenesis of matrix vesicles induced calcification of recipient vascular smooth muscle cells
Supplemental material, sj-tif-4-vmj-10.1177_1358863X211024721 for Regulation of reactive oxygen species in the pathogenesis of matrix vesicles induced calcification of recipient vascular smooth muscle cells by Neal X Chen, Kalisha D O’Neill, James M Dominguez and Sharon M Moe in Vascular Medicine
Supplemental Material
sj-tif-5-vmj-10.1177_1358863X211024721 – Supplemental material for Regulation of reactive oxygen species in the pathogenesis of matrix vesicles induced calcification of recipient vascular smooth muscle cells
Supplemental material, sj-tif-5-vmj-10.1177_1358863X211024721 for Regulation of reactive oxygen species in the pathogenesis of matrix vesicles induced calcification of recipient vascular smooth muscle cells by Neal X Chen, Kalisha D O’Neill, James M Dominguez and Sharon M Moe in Vascular Medicine
Supplemental Material
sj-tif-6-vmj-10.1177_1358863X211024721 – Supplemental material for Regulation of reactive oxygen species in the pathogenesis of matrix vesicles induced calcification of recipient vascular smooth muscle cells
Supplemental material, sj-tif-6-vmj-10.1177_1358863X211024721 for Regulation of reactive oxygen species in the pathogenesis of matrix vesicles induced calcification of recipient vascular smooth muscle cells by Neal X Chen, Kalisha D O’Neill, James M Dominguez and Sharon M Moe in Vascular Medicine
Footnotes
Acknowledgements
We are grateful to the assistance of Gosia Kamocka (Indiana Center for Biologic Microscopy, Indiana University School of Medicine, Indianapolis, IN) for assistance with imaging.
Declaration of conflicting interests
The author(s) declared the following potential conflicts of interest with respect to the research, authorship, and/or publication of this article: Dr Moe is a scientific consultant for Amgen, Sanifit, and Ardelyx and receives grant support from the VA, NIH, Chugai Pharmaceuticals, and Keryx. The other authors have nothing to disclose.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was funded by a Veterans Administration Merit Award from United States (US) Department of Veterans Affairs IBI01BX001471, Biomedical Laboratory Research and Development Service (SMM), and the NIH O’Brien Center P30-DK079312.
Supplementary material
The supplementary material is available online with the article.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.