
Abstract
Abdominal aortic aneurysm (AAA) is a common disease associated with significant cardiovascular morbidity and mortality. Up to now, there is still controversy on the choice of treatment method of AAA. Even so, the mechanisms of AAA progression are poorly defined, making targeting new therapies problematic. Current evidence favors an interaction of the hemodynamic microenvironment with local and systemic immune responses. In this review, we aim to provide an update of mechanisms in AAA progression, involving hemodynamics, perivascular adipose tissue, adventitial fibroblasts, vasa vasorum remodeling, intraluminal thrombus, and distribution of macrophage subtypes.
Keywords
Introduction
Abdominal aortic aneurysm (AAA), caused by kinds of exogenous and endogenous factors, is a pathologic local dilation of the abdominal aorta, which is associated with significant cardiovascular morbidity and mortality. Most patients with AAA usually have no symptoms until the aneurysm rupture. But once the aneurysm ruptures, the overall mortality rate is up to 80%. 1 With the aging of the population, its incidence increases year by year. Over the last decade, endovascular aneurysm repair (EVAR) has rapidly become the mainstream treatment option of the world due to a significant reduction in 30-day operative mortality and length of hospital stay when comparing with open surgical repair; however, with longer follow-up, more patients with local complications require additional intervention.2–4 Even though the need for better therapies is so urgent, there is still no other clinically identified or validated treatment that can prevent the progression and rupture of AAA. Therefore, it is very worthwhile to go into the mechanisms of AAA. In this paper, we aim to provide an update of mechanisms in AAA progression.
Inside-out inflammation theory and hemodynamics
Controversies of AAA animal model
Currently, the research on pathophysiological mechanisms of the occurrence and development of human AAA is mainly based on studies in animal models. However, due to the influence of various objective reasons, such as breeding cycle, feasibility, and other factors, current animal models of AAA are primarily rodents, 5 such as rats and mice, which were treated mainly by chemical induction and/or gene knockout techniques, including subcutaneous infusion of the angiotensin II-induced apolipoprotein E knockout (ApoE–/–) mice model, 6 and intra-aortic perfusion of elastin-induced 7 and abluminal application of calcium chloride (CaCl2)-induced8,9 rodent models. They can also be divided into three types according to their processing mechanisms: (1) collagen elastase diseases that cause AAA: intra-aortic perfusion or abluminal application of elastase 10 with or without oral beta-amino acrylic nitrile (BAPN)11,12 or systematic blocking of the activity of transforming growth factor-beta (TGF-β)13,14; (2) atherosclerosis causing AAA (most common in humans): ApoE–/– or low-density lipoprotein receptor knockout (LDLr–/–) mice 15 with or without angiotensin-II (Ang II) subcutaneous infusion; and (3) inflammatory AAA (least common): abluminal application of CaCl2.
However, the representation of these popular animal models is controversial. Ang II is commonly applied as the low natural incidence of aortic aneurysm in mice with severe atherosclerosis (ApoE–/–); 16 however, the AAAs induced by Ang II present the accumulation of macrophages, degradation of elastin, intermural hematoma, and thrombi in the media of the suprarenal aorta, 17 which is more similar to the pathophysiology of aortic dissections or aortic dissecting aneurysms. Application of CaCl28,9 induces damage to the medial membrane and endothelium accompanied by sedimentation of calcium salts in the elastic lamina and calcification. However, the low incidence of aneurysm formation, extensive calcium salt deposition, especially the absence of matrix metalloproteinases (MMP)-2 and -9 in the aneurysm wall, 18 suggest that it is widely different from common human AAA. Similarly, extraluminal elastase application-induced AAA also lacks some characteristics of common AAAs in humans, such as medical angiogenesis and non-neutrophilic infiltration. 10 Intra-aortic perfusion of elastin 7 can cause extensive damage to the elastic lamina and extracellular matrix in the adventitia, which has been widely used in the mechanism study because of its similar pathological characteristics with human AAA, but its self-healing tendency 19 indicates the theoretical insufficiency. Moreover, in addition to the above characteristics, all of the three popular models were criticized for the low degree of expansion (< 70–100%), no obvious intraluminal thrombus, and poor reproducibility in nonrodent models in the absence of BAPN or anti-TGF-β antibodies. 20 BAPN is known to induce abnormal cross-linking of aortic vascular matrix collagen, which leads to aortic dissection and intramural hematoma. 21 Recently, Lu et al. 11 reported the development of a chronic model of AAA in mice with intraluminal thrombus after a combination of extraluminal elastase application and continuous oral BAPN; however, the authors provided no evidence for the absence of medial dissection and it remains an animal model of dissecting aneurysm but not AAA. Systemic blocking TGF-β activity can impair vascular tissue repair capacity by systemic regulation of macrophages, which can induce self-limited dilation to persistent aortic dilation.13,14 But it might be a victim of its success, as the abnormal systemic immune conditions caused by systemic blocking TGF-β activity are far-fetched to mimic the systemic immune status of patients with AAA. It was also reported that there was no correlation between the human TGF-β receptor gene and AAA. 22 Accordingly, animal models of AAA induced by anti-TGF-β antibodies might not be reliable for the drug research and development of AAA.
It is also necessary to note that some investigators are not using ultrasound to show AAA development in animals but are relying on measurements of fixed tissue, which can be misleading, as it was reported that the increased aortic dilation measured by anatomy in the sham group was even more than 50% (51.9 ± 29.2%) compared with baseline. 12
Human AAA histopathology: hints of the inside-out inflammation theory
Degradation of the elastic lamina and medial attenuation are the typical pathological features of human AAA specimens. 23 But evidence from animal experiments 24 and clinical experience of endarterectomy suggests that the probability of sustained aortic dilation caused by damage to the elastic lamina and the media is very low. 25 The tensile property of the vascular wall is mainly derived from collagen fibers in the stroma, and the elastic fibers, with very low tensile strength, are mainly to maintain the elasticity of the vascular wall and preserve elastic potential energy. 24 Therefore, the expansion of aneurysms seems to be mainly due to the changes in collagen fibers in the vascular matrix. 24 On an electron microscope, the spatial arrangement of collagen fibers in human AAA adventitia whose thickness is not significantly reduced is no longer a parallel structure like a multilayer stent, but a disorderly deposition structure where the collagen cannot exert its tensile properties, and it was also found that in the early stage of AAA (small AAA) the elastic lamina layer has already been severely degraded.25,26 Therefore, the degradation and remodeling of collagen fibers in the media and adventitia of the aortic wall seem to be the major contributor to AAA occurrence and progression. However, the origins of the sustained inflammation causing the degradation and remodeling of vessel walls are still unknown. Does it follow an inside-out pattern or an outside-in one?
Two animal models of AAA induced by different elastase treatments seem to provide a reasonable answer. In 2016, Busch et al. 10 reported the histological differences between intraluminal elastase perfusion (PPE group) and extraluminal elastase application (EPA group). The results suggested that the intraluminal elastase perfusion models showed the destruction of elastic layers followed by chronic inflammation, as well as medial angiogenesis, a key feature in human AAA; whereas the extraluminal elastase application-induced aneurysms preserved endothelial cell function and elastic fibers but showed ongoing acute inflammation, mainly in the adventitia. Therefore, the inside-out inflammatory response induced by intraluminal elastase perfusion seems to be more in line with the human AAA pathophysiological mechanisms, whereas the EPA group, presenting acute neutrophils infiltrating to the adventitia, seems to be more similar with a human inflammatory AAA which has the same pathological feature. 27
New animal models and hemodynamics
The intraluminal elastase perfusion model suggests the possible mechanism of AAA development; however, the limitations of the model, such as low expansion rate and self-healing tendency, make it unconvincing to explain the mechanism of AAA progression. Hemodynamics theory, which has been studied for the long-term in the vascular field, seems to explain this. In 2011, Mata et al. 20 reported a new animal model of AAA by combining two potential causes of metalloproteinase secretion: inflammation and turbulent blood flow. Via stenosis induced by a partial delegation at the infrarenal aorta and a traumatic injury to the external aortic wall around the ligation, 60–70% of the animals developed AAAs by day 3, and the AAAs reached a dilation ratio of more than 300%, whereas in the stenosis group, injury group, and sham group, obvious aortic dilatation was not found. The histopathological analysis showed obvious inflammatory infiltrate, mesenchymal proliferation, medial neo-angiogenesis, elastin laminal degradation, collagen deposition of adventitia, and a significant increase of MMP-2 and -9. 20 This is also the only one of many AAA models whose diameter can expand to 300% and whose intraluminal thrombus is as obvious as that of human AAA, suggesting that hemodynamics may be of great importance in the occurrence and progression of AAA.
At present, the studies of hemodynamics mainly focus on atherosclerosis, including perturbed flow, oxidative stress, and endothelial mechano-sensors. First, the perturbed flow impacts vascular endothelial function. Based on the development of animal models of disturbed flow, steady and laminar flow is believed to reduce flow resistance and induce pro-survival signals which can maintain endothelial cell survival and increase endothelial barrier function, whereas the perturbed flow with low shear stress will expose the endothelial cells to the higher shear stress gradients and decrease the pro-survival signals, leading to intensive monocyte attachment, lipid accumulation in the vascular wall, and enhanced atherosclerosis progression. 28 Second, perturbed flow induces oxidative stress. Continuous turbulence flow will cause sustained activation of pro-oxidant mechanisms resulting in redox-sensitive gene expression in endothelial cells, yet steady and laminar flow will initially activate pro-oxidant mechanisms, but also induces compensatory antioxidant defenses. Constantly exposed to turbulence, endothelial cells will lose the compensatory mechanism and expand the proinflammatory properties, inducing oxidative stress and corresponding endothelial dysfunction, even endothelial cell autophagy and apoptosis. 29 Finally, mechano-sensors, a mechanism by which endothelial cells can transform the mechanical shear force felt on the cell surface into intracellular biochemical signals, would be destroyed by the perturbed flow. These mechanical sensors are very important for endothelial cells to inhibit leukocyte adhesion, platelet adhesion, mediating shear stress-dependent endothelial NO release, and other vascular protective mechanisms; however, these receptors can be impacted and even stripped by hyperlipidemia, turbulence, and other factors, thus losing the endothelial protective characteristics. 29
Hemodynamics also plays an important role in regulating the synthesis of the vessel matrix. Hosseini et al. 30 developed a flow device that can expose 3D engineered vascular smooth muscle cells (VSMCs) to different patten of flow conditions to study the relationship between extracellular matrix remodeling and hemodynamics. It was found that the synthesis of the extracellular matrix was not affected by hemodynamics, although the production of MMPs and its inhibitor TIMP1 were significantly affected by hemodynamics. When in a high-velocity pulsatile flow, TIMP1 secretion and elastin synthesis tended to advantage; in a low-velocity disturbed flow condition, collagen synthesis and elastin degradation tend to advantage, and the TIMP1 synthesis decreases.
Above all, hemodynamic plays an important role in vessel inflammation and matrix remodeling, but nowadays its studies on AAA mostly remain in computer modeling and analysis, more often limited to physical forms. 31 Turbulence and low shear stress caused by the special spatial structure of AAAs and their effect on the AAA progression remains to be further researched.
Outside-in inflammation theory and perivascular adipose tissue
The outside-in inflammation theory and animal models
As promising as the new hemodynamic animal model may be, it does not mean that outside-in inflammation theory is good for nothing. The sustained inflammatory cell infiltration and matrix remodeling of the outer membrane combining with a relatively intact intima in the early stage of human AAA indicate the rationality of the outside-in inflammation theory. 23 The study of Mata et al. 20 also showed that only aortic stenosis without the injury of the outer layer could not result in obvious arterial dilatation, indicating the presence of inflammation from the outside in. In recent years, the research frontiers of this theory mainly focus on the perivascular adipose tissue (PVAT).32,33
Perivascular adipose tissue with AAA
There is no clear boundary between the adipose tissue around the human great vessels and the adventitia, and the adipose tissue around the microvessels has even become a part of the vascular wall; therefore, in recent years, the perivascular adipose tissue is referred to as a component of adventitia. 34 Broadly, PVAT includes the connective tissue structure beyond the elastic lamina of the vascular wall, including vasa vasorum and kinds of cells like fibroblasts, lymphocytes, adipocytes, etc.33,34 Vascular homeostasis can be regulated by cytokines or chemokines secreted from adipocytes or other stromal cells such as fibroblasts, macrophages, and lymphocytes because of the close connection between PVAT and vascular wall.33,34
Recent evidence highlights the role of PVAT in the development of inflammation. It is reported that the chemotactic factor for monocyte (MCP)-1 expressed by adipocytes around the coronary arteries of patients with coronary artery disease is 50 times more than that of other adipose tissues such as subcutaneous or visceral adipose tissues in the same individual. 35 Horimatsu et al. 36 transplanted 50 mg of abdominal aortic PVAT isolated from high-fat diet-fed wild-type mice onto the abdominal aorta of lean, low-density lipoprotein receptor (LDLr) knockout mice and found that the thoracic aortic atherosclerosis increased, combining elevated MCP-1, tumor necrosis factor-alpha (TNF-α), and reduced adiponectin, suggesting augmented inflammation as a potential mechanism for the remote vascular effects of transplanted PVAT.
PVAT is also likely to play an important role in the development of AAA. Kugo et al. 37 reported that in the AAA model induced by hypoperfusion, the AAA diameter, mesenchymal stem cells, and adipocytes significantly decreased in the PVAT-removed vascular wall compared with that in the vascular wall with PVAT. Liu et al. 38 and Sakaue et al. 39 reported the PVAT also plays an important role in promoting the formation and progression of AAA in Ang II-induced AAA models. A recent mechanism study 40 using high-throughput sequencing technology to identify the relationship between PVAT of human AAA and the inflammation associated with congenital and acquired immune system, found that both innate and adaptive immune-response genes along with genes related to cell apoptosis pathways, the metabolic process of collagen and extracellular matrix degradation were strongly overrepresented in PVAT of AAA compared with PVAT of the not-dilated aorta, indicating the PVAT dysfunction of human AAA patients. The dysfunction can also be proved by the study of Piacentini et al., 40 who found that a large number of T cells were infiltrating in PVAT of human AAA, and the infiltration degree was significantly correlated with AAA diameter.
Of course, PVAT is not all bad. A healthy PVAT can also secrete a mass of anti-inflammatory factors unless adverse factors such as smoking and hyperlipidemia tilt the scales towards the proinflammatory side.32,33 It was reported that when PVAT was removed from the LDLr knockout mice, non-high-fat feeding can cause endothelial dysfunction and aggravation of atherosclerosis. 41 Terada et al. 42 transplanted the thoracic aorta PVAT of wild-type mice to the abdominal aorta of ApoE–/– mice and found that the area of atherosclerosis plaque in the transplanted mice was significantly lower than that of the sham group, which was mediated by endocrine mode of PVAT and the effect of type 2 (M2) macrophages. Though it was found that obesity is significantly related to the occurrence of human AAA, whether obesity is related to the AAA growth remains controversial.43,44
Vascular fibroblasts and inflammation
Vascular fibroblasts are an important cell type in PVAT and are even called the ‘whistleblowers’ of vascular inflammation. 45 As early as 1996, it was reported in Circulation that the proliferation of fibroblasts in PVAT of the balloon-injured coronary artery was earlier than that of medial VSMCs, and the proliferation activity was also greater than that of VSMCs during the whole process of neointima formation, which was first presented as the theory of adventitial remodeling. 46 Fibroblasts also play an important role in innate and specific immune responses. 45 In vascular inflammatory reaction, activated fibroblasts proliferate and migrate to the inner layer, and induce the phenotypic transformation of VSMCs from contractile type to secretory type, participating in the regulation of extracellular matrix. Liu et al. 47 reported that adventitial fibroblasts of Ang II infusion mice could be strongly activated by hyperhomocysteinemia, which was significantly related to human AAA (odds ratio [OR] 7.39; 95% CI: 3.71–14.72). The activated fibroblasts produced a large amount of proinflammatory cytokine and chemokines such as IL-6 and MCP-1 via induction of oxidative stress, promoting the recruitment and infiltration of monocyte/macrophage, which produced a lot of MMPs to degrade extracellular matrix and participate in the development of AAA. Tilson et al. 48 found that the MCPs and derivative neutrophil chemokines expression of adventitial fibroblasts was significantly greater than that of dermal fibroblasts by tagging cDNA probe technology. Gäbel et al. 49 found that the significantly up-regulated expressing factors of end-stage AAA were converged at the signal pathway of hypoxia-inducing factor-1α (HIF-1α), and these factors were significantly overexpressed in the adventitial fibroblasts rather than infiltrating inflammatory cells, suggesting that adventitial fibroblasts play an important role in the procession of AAA.
In addition to the proinflammatory properties, adventitial fibroblasts may also play an important role in the AAA repair. It was reported that TGF-β could induce adventitial fibroblasts to produce type I and III collagen, which were responsible for the load-bearing characteristics and ductility. Both types of collagen are of great significance for AAA growth and rupture. For example, vascular Ehlers–Danlos syndrome, which is prone to aortic rupture, is known for a lack of type III collagen. Yang et al. 50 reported that the adventitial fibroblasts in the CaCl2-induced animal models could be divided into two major subpopulations. Fibro-1 highly expressed Col1a1, Col1a2, and Col3a1, suggesting this cell population is important in collagen synthesis, whereas Fibro-2 highly expressed AAA-related genes and cell cycle genes, suggesting that the Fibro-2 population might be inflammatory and actively proliferating.
Vasa vasorum (VV) intimal hyperplasia and the hypoxia theory
In recent years, a novel ‘hypoxia theory’ on AAA has been proposed, 51 believing that AAA might be originated from the hypoperfusion of the adventitial vasa vasorum, which leads to aortic wall hypoxia, extracellular matrix damage, continuous sac dilation, and even AAA rupture. Tanaka et al. 51 studied human AAA specimens and found that even though the vasa vasorum (VV) of the AAA sac proliferated obviously, HIF-1α was positive in all the layers of the sac wall, which corresponded to the areas with weak Heme B signals (a specific marker for blood stock and may reflect the level of tissue blood flow). The study also revealed marked intimal hyperplasia in the adventitial VV of the AAA sac, with a significantly smaller luminal area compared with that in the non-dilated neck adventitia or the control specimens. Moreover, this intimal hyperplasia is most likely due to VSMC proliferation rather than atherosclerotic plaque. 51
The classic AAA animal model supporting the hypoxia theory is the rat model established by Tanaka et al.,52,53 which was created with a combination of polyurethane catheter insertion and suture ligation of the infrarenal abdominal aorta. It was proved that the morphological and pathological characteristics of the new model were similar to human AAA, including intimal hyperplasia of adventitial VV and distribution of HIF-1α and Heme B. 52 Kugo et al. 54 studied the hypoperfusion-induced animal model and showed that with the dilation of the AAA sac, collagen and elastin levels of the sac wall were significantly reduced. This is different from the phenomenon of decreased elastin and large deposition of collagen in human AAA specimens, 25 suggesting that wall hypoxia might be an important factor of AAA progression mechanisms rather than the whole story. It also found that HIF-1α, ET-1, Ang, MMP-2/-9, and MCP-1 were all colocalized with α-SMA+ cells in the first 3 hours of hypoxia but not in Mac387+ cells, which were colocalized 6 hours later, 54 suggesting hypoxia affected the contraction of VSMC function and activity and might be a possible inducer of the phenotypic transformation of VSMCs in the early stage of AAA.
However, it is not clear whether VV stenosis occurred before degeneration and dilation of the aortic wall or resulted from the changes occurring during the AAA progression. It seems to be suggestive that the changes of VV in hyperlipidemia animal models were detected well before plaque formation and even preceding the onset of endothelial dysfunction.55,56 The VSMC phenotype transformation also may promote some indications. Ailawadi et al. 57 found that the phenotype switch of VSMCs probably occurred before the formation of AAA based on the elastase perfusion model. Moehle et al. 58 reported that MCP-1 secreted from bone marrow-derived inflammatory cells seemed to be the key to the phenotype switch of VSMCs in the AAA mice model induced by elastase perfusion. As mentioned above, adventitial fibroblasts and adipocytes of abnormal PVAT could secrete plenty of MCP-1.35,48 Since the abnormal hemodynamic pattern is unlikely to be established before aortic dilation, it is reasonable to assume that the aortic wall hypoxia might be the major trigger of MCP-1 in PVAT, which is in line with Kugo’s study that hypoxia affected the contraction of VSMC function and activity and may be a possible inducer of the phenotypic transformation of VSMCs in the early stage of AAA. 54 In other words, the VV stenosis might be prior to the VSMC phenotypic transformation and AAA formation. What is more, the hypoxia-induced AAA model expanded significantly within only 14 days after modeling and the sac rupture occurrence reached 20% within 28 days,52,53 suggesting that adventitial VV hypoperfusion and sac wall hypoxia are more likely to be the crucial mechanisms of terminal AAAs. Overall, adventitial VV remodeling and hypoxia theory are likely to run through the whole process of AAA development and progression, which is well worth investigating further.
Intraluminal thrombus and distribution of macrophage subtypes: view of immunology
The hypothesis of autoimmunity and autoimmune disease
Since Beckman proposed the hypothesis of autoimmunity on the etiology of AAA in the 1980s, 59 the autoimmune disease views of AAA have always been considered. 60 Autoimmune abdominal aortic protein 40 (AAAP-40), which once had been paid much attention, was finally proved to be a normal tissue structural protein targeting the major arteries of the whole human body.60,61 Since then, the voice of autoimmune disease on AAA gradually decreased but never disappeared. Lu et al. 62 reported that lesions from AAA patients contained oligoclonal populations of T cells with multiple identical copies of β-chain T cell receptor (TCR) transcripts, believing that AAA is a specific Ag-driven autoimmune T cell disease. However, recent research does not support this hypothesis. Menges et al. 63 reported that the secondary expanded aneurysm wall after EVAR showed significant thinning of the intima–media layer accompanied by a scarcity of cells, with only a few macrophages and smooth muscle cells left, and the lymphocytes that are commonly seen in AAA adventitia almost disappeared in the wall of secondary expansion. It seems to contradict the hypothesis of autoimmune disease, as the EVAR surgery, at least in theory, is impossible to alter the human immune status.
In recent years, views on autoimmunity have attracted more attention due to the regulatory T lymphocytes (Treg). The main opinion is that AAA progression is at least partly caused by the reduction of Tregs in the circulation of AAA patients, considering that Tregs have a potential effect of inhibiting the disease aggregation.64,65 Suh et al. 65 reported that the proportion of Tregs is decreased in AAA patients compared with matched control patients with significant vascular disease. They also found that human CD4+ T cells were able to drive aneurysm formation in Rag–/– mice (which have no mature T and B lymphocytes and are resistant to CaCl2 aneurysm induction), whereas ex vivo augmentation of human Tregs resulted in decreased aneurysm progression. Ait-Oufella et al. 64 reported that Tregs could notably reduce the sac wall infiltration of macrophages and CD4+ T lymphocytes, preserve medial VSMCs, and bring down the levels of proinflammatory factors and MMPs, which could significantly inhibit the aneurysm formation and rupture in the Ang II-induced AAA model.
Intraluminal thrombus and macrophage distribution: a defective armor
At present, studies on intraluminal thrombus (ILT) remain great gaps for improvement. Lindholt et al. 66 reported that low-dose aspirin could significantly reduce the growth rate and rupture risk of AAA measured 40–49 mm (p = 0.0173) but had no effect on that of AAA smaller than 40 mm (p = 0.3447). Elbadawi et al. 67 queried the National Inpatient Sample (NIS) database (2009–2016) and found that use of antiplatelet drugs was associated with a lower rate of AAA (OR = 0.7; 95% CI: 0.68–0.72, p < 0.001), AAA dissection (OR = 0.53; 95% CI: 0.50–0.57, p < 0.001), and AAA rupture (OR = 0.67; 95% CI: 0.54–0.82, p < 0.001). Though there are some inconsistent studies, 68 nevertheless, given the antithrombotic effect of aspirin and higher ILT tendency of larger AAAs, 69 the presence of ILT might play an adverse effect on the aneurysm sac wall. Vorp et al. 70 suggested that the thicker ILT in AAA induced localized hypoxia, leading to increased, localized mural neovascularization and inflammation, as well as regional wall weakening. The hemodynamic study using finite element analysis showed that a larger thrombus size in AAA was associated with lower stress, but also with a higher AAA growth rate.71–73 The average ILT thickness of ruptured AAA with a smaller diameter was also significantly higher than that of unruptured AAA with a smaller diameter or larger diameter, 74 suggesting that ILT plays a more imminent role in the process of AAA growth than the stress acting on the wall. Nonetheless, is the mechanism real due to hypoxia suggested by Vorp?
Wiernicki et al. 75 and Folkesson et al. 76 did further studies on ILT and AAA progression and found that the expression of MMP-9 and -8 in ILT drastically decreased from the lumen-side to the wall-side, and the expression of MMPs in the walls with thick ILT (removed) markedly reduced compared with that in the walls with thin ILT (removed) or without ILT, whereas the expression of its inhibitor TIMP1 increased. Kazi et al. 77 reported that the expression of MMPs in the non-ILT walls of AAA was more than three times higher than that in the walls with ILT, and the macrophages, the only stroma cell related to MMPs, were mainly concentrated in the non-ILT walls (p < 0.05), though there was no difference between distribution of VSMCs and T cells. This raises a new question of how can ILT weaken the wall while playing a protective role in reducing the infiltration of macrophages and expression of MMPs in ILT locations?
Immunology seems to be the key to solve the problem. Classically activated macrophages (M1) tend to promote tissue injury, whereas alternatively activated macrophages (M2) often tend to anti-inflammation, wound repair, and fibrosis, in a mechanism of secreting TGF-β, vascular endothelial growth factor (VEGF), epidermal growth factor (EGF), and other factors to promote collagen synthesis and deposition with stromal cells such as VSMCs and fibroblasts. 78 Th2 cytokines, interleukin (IL)-4, and IL-10, dominant in the AAA walls, 79 are the main factors that trigger M2 activation in infiltrated macrophages. 78 Based on the animal experiments, 17 it has been proved that in the early stage when AAA formed (28 days), the M1 phenotype was dominant in AAA walls, indicating that the aneurysm walls were infiltrated by proinflammatory cells. Later, after the AAA formation (56 days), the amount of M2 phenotype and M2/M1 ratio were significantly increased, suggesting that compensatory repair was the main histological reaction after AAA formation. Dutertre et al. 80 reported that the M2 phenotype was significantly superior to M1 in the media and adventitia of human AAA. But Boytard et al. 81 reported opposite results that macrophages could hardly be seen in the media and M1 was mainly concentrated in the adventitia whereas M2 was mostly in the luminal side of ILT. Note that the most important thing here was all 20 samples in this study contained thick ILT. 81 As macrophages seem to be more accumulated in ILT-free sites of human AAA, 82 it is speculated that ILT could impact the distribution of macrophage subtypes, and keep M2 macrophages from repairing the wall, eventually leading to the relatively upregulated matrix degradation and making the AAA easy to rupture.
At present, much research focuses on the effects of M1/M2 balance on AAA progress. Dale et al. 83 reported that a preinjection of M2 macrophages into the tail vein of the CaCl2-application mice model had an inhibitory effect on AAA diameter, suggesting the balance of M1/M2 had a direct regulation on the progression of AAA. Chen et al. 84 analyzed the genome data of human AAA patients and found that the incidence of AAA in women was lower than that in men (about 1:4), and may be due to the higher estrogen level which promoted the M2 phenotype transformation of macrophages. Feldmann et al. 15 reported that dabigatran inhibited the progression of atherosclerosis in LDLr–/– mice by reducing the proinflammatory cytokines and accumulation of M1 macrophages in the aortic wall and PVAT. It is also reported that rivaroxaban, an oral factor Xa inhibitor that has been proved to be able to attenuate both Ang II- and CaCl2-induced AAA progressions, 85 reduced MMP9 expression and exerted anti-inflammatory and anti-oxidative stress properties in incubated human AAA tissues.85,86
In summary, the infiltrating macrophages in the AAA walls play an important role in both pathologic and compensatory mechanisms in the progression of human AAA, especially in the remodeling of extracellular matrix and the regulation of vessel inflammation. 87 When thick ILT is formed, it may cause a significant reduction of local infiltration of M2 phenotype macrophages which had a compensation mechanism of tissue repair, even though the MMP expression is lower in thick ILT walls than in thin ILT and non-ILT walls. Combined with the change of Th1/Th2 balance (T helper cells decrease and cytotoxic T cells increase) and VSMCs apoptosis 82 when thick ILT formed, the loss of M2 would worsen the tensile strength of the aortic wall, accelerate AAA progression, and increase the risk of rupture.
Conclusion and perspective
At present, the treatment of AAA, especially EVAR surgery, is still confined to morphological repair. To better slow down AAA progression and prevent rupture, more attention should be paid to the mechanism study of AAA progression. Recent evidence highlights the role of hemodynamics, PVAT, and macrophages subtype balance in the AAA progression (Figure 1). How to reduce the influence of blood flow to the aneurysm wall and reverse the proinflammatory property of PVAT is particularly important for the treatment of AAA. For AAA with ILT, how to predict the progression trend, rupture risk and possible rupture site, and whether to take more aggressive or conservative action need further exploration. Given the influence on AAA progression, the macrophage system, especially M1/M2 balance, might become a promising target for AAA diagnosis, prediction, and treatment in the future.87,88
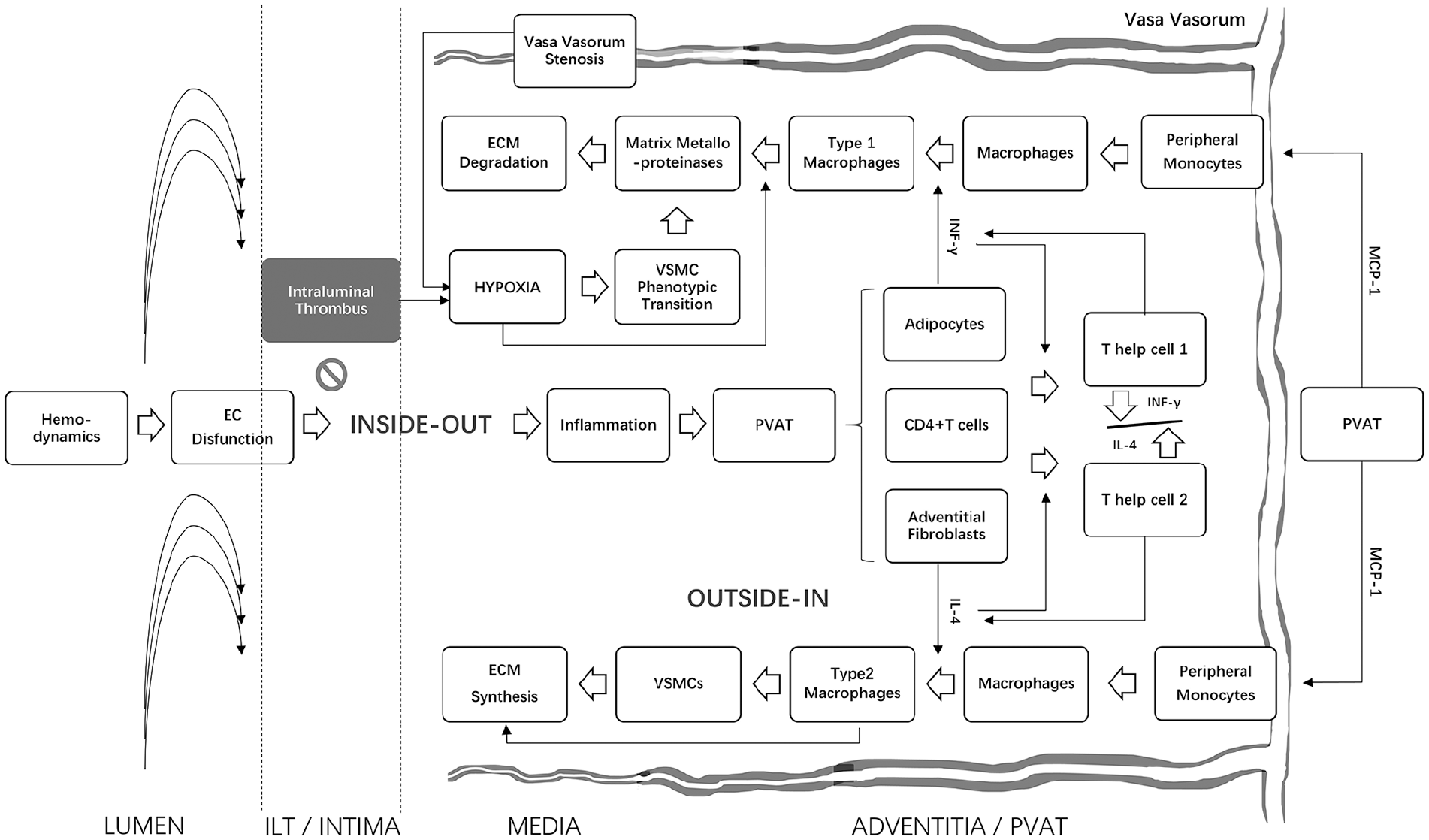
Interactions of hemodynamics, PVAT, vasa vasorum, intraluminal thrombus, and macrophage subtypes in AAA progression. The figure provides an overview of potential mechanisms involved in AAA progression. Inflammation is driven by hemodynamics and amplified in PVAT. The adventitial fibroblasts produce a mass of MCP-1 to accumulate monocytes and promote macrophage infiltration. Meanwhile, the inflammatory cells caused infiltration of CD4+ T cells that would transform into Th1 type and Th2 type. Th1 would secrete cytokines such as INF-γ to induce M1 into having the properties of proinflammation and matrix degradation, and Th2 would secrete cytokines such as IL-4 and -13 to induce M2 into having the properties of anti-inflammation and pro-fibrosis. Heavy ILT may inhibit the inside-out inflammatory pathway and cause a significant reduction of local infiltration of M1 and M2 macrophages, thereby inhibiting the compensation mechanism of tissue repair. Since the inferior mesenteric and lumbar arteries are often narrowed or occluded by heavy ILT, ILT would worsen the VV hypoperfusion and exacerbate aneurysm wall hypoxia, which may lead to more severe matrix degradation, accelerate AAA progression, and increase the risk of rupture.
Footnotes
Declaration of conflicting interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors received no financial support for the research, authorship, and/or publication of this article.