
Abstract
Despite being recognised over a century ago, the aetiology and pathogenesis of large vessel vasculitis (LVV) still remains elusive. Takayasu’s arteritis (TA) and giant cell arteritis (GCA) represent the two major categories of LVV, each with distinctive clinical features. Over the last 10 years an increased understanding of the immunopathogenesis of the inflammatory cascade within the aortic wall has revived the view that LVVs may represent subtypes of the same pathological process, with implications in the treatment of this disease. In this review, the histological, genetic and immunopathological features of TA and GCA will be discussed and the evidence for a common underlying disease mechanism examined. Novel markers of disease activity and therapies based on advances in our understanding of the immunopathogenesis of these conditions will also be discussed.
Keywords
Introduction
Over recent years considerable advances into the immunological and genetic pathogenesis of large vessel vasculitis (LVV) have been made. This has paved the way to understanding the natural history of the disease, identifying novel biomarkers for the diagnosis and prognosis of disease progress and personalisation of immunomodulatory treatments for inducing and maintaining remission of the disease. However, the aetiology and pathogenesis of LVV remains poorly understood and still many controversies exist regarding the pathological identities of LVVs, namely Takayasu’s arteritis (TA) and giant cell arteritis (GCA) (Figure 1). Despite commonality in the immunopathogenesis of inflammation of the aortic wall, each condition presents significantly different clinical features. In this review we will discuss the histological, genetic, and immunopathological characteristics of these conditions in an attempt to clarify this issue as well as summarise recent advances and current controversies. The main features of both conditions are summarised in Table 1.
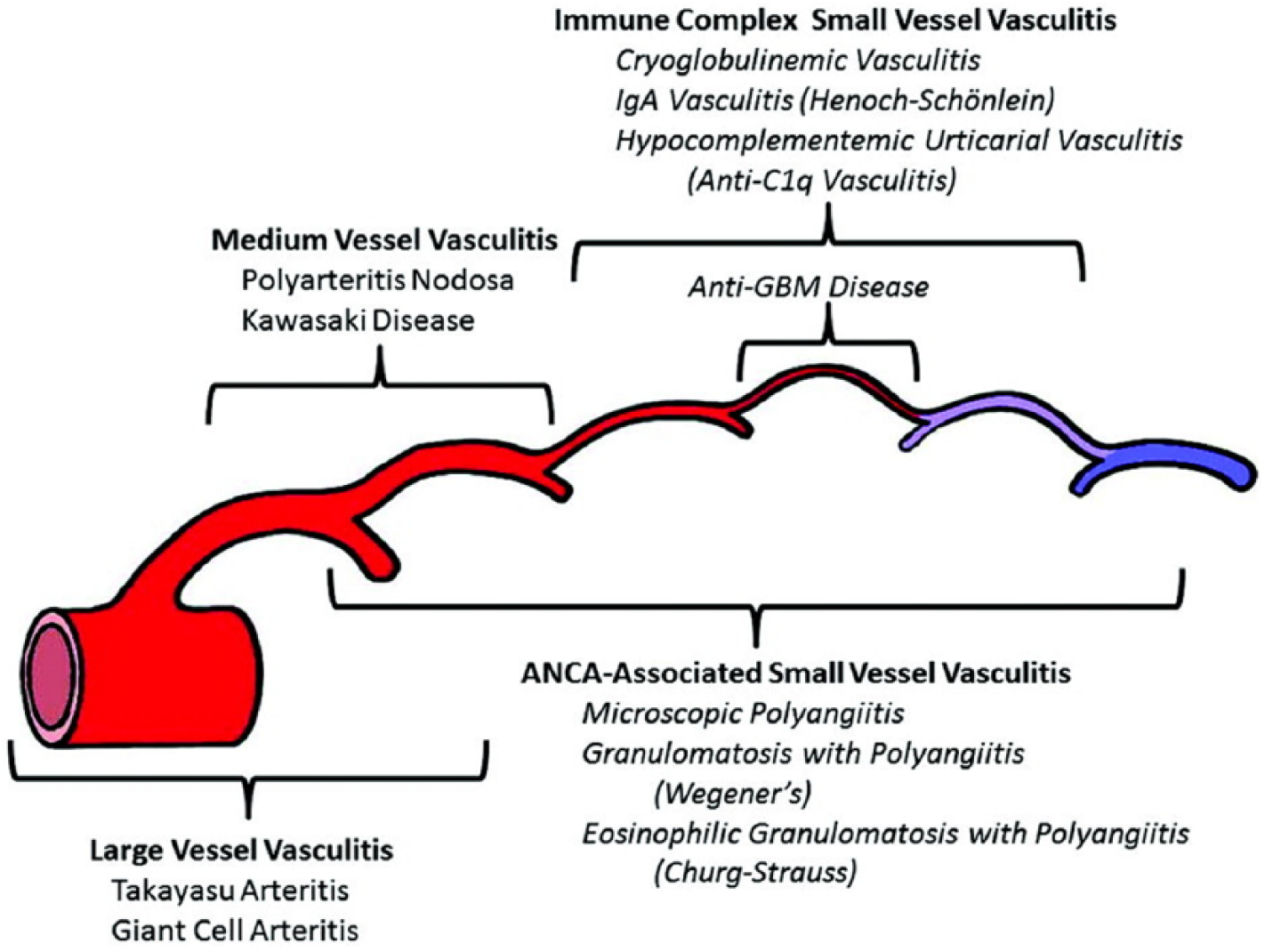
Chapel Hill Consensus Conference Nomenclature of Vasculitides, which displays they distribution of vessel involvement by large vessel vasculitis, medium vessel vasculitis, and small vessel vasculitis. Reproduced with permission from John Wiley & Sons. 33
A comparison of the significant characteristics of Takayasu’s arteritis and giant cell arteritis.

HLA, human leukocyte antigen; IL, interleukin.
Disease classification
TA, although first identified in 1908 by the Japanese ophthalmologist Mikito Takayasu who described a ‘case of peculiar changes in the central retinal vessels’, significance in terms of large vessel pathology was not fully comprehended until 1940, when the ophthalmic lesions were associated with panarteritis of the aorta at autopsy. 1 In contrast, GCA is a relatively new definition in terms of classification and diagnosis. Owing to its comorbidity with polymyalgia rheumatica, GCA is now classified as a distinct inflammatory condition and is the most common primary vasculitis in adults.
Epidemiology
TA is a rare disease affecting primarily young Asian women, with a female to male ratio of 8–9:1 in Japan. 2 Estimates from western countries have shown incidences of 0.4–2.6 per million population.3–6 Evidence for a genetic basis was provided by the observation that the vast majority of TA cases originate from Japan, with a reported 100 times higher incidence in Eastern Asia compared to Europe and the United States, and with a peak age of onset at 15–30 years of age.7,8
The true incidence of GCA is unknown, with incidences ranging from 0.49 to 27.3 per 100,000 in the USA. 9 An incidence of 17.8 per 100,000 has been reported in individuals over 50 years of age. 10 The main difference in presentation between the two conditions is the age of disease onset, with GCA rarely occurring before age 50 11 and having a peak incidence at 60–80 years of age. 12 Geographical variation is opposite to TA in that it is more common in northern Europeans. 13 Although both TA and GCA display a strong predilection for women, 14 GCA is usually diagnosed just before or after the menopause as opposed to TA where patients are of child bearing age. 15
Histopathology of Takayasu’s arteritis (TA) and giant cell arteritis (GCA)
TA and GCA are characterised by a chronic granulomatous inflammatory reaction of large vessels. 16 However, there are also histological differences in each condition. In TA, the characteristic feature is the strong fibrotic reaction seen in the aortic adventitia, which is in excess of that seen in any other aortitis.17,18 Microscopically, active TA can be divided into an acute inflammatory phase and a chronic fibrotic phase. In the acute phase, an inflammatory infiltrate consisting of lymphocytes and plasma cells is primarily seen around the vasa vasorum within the adventitia, and is later infiltrated with multinucleated giant cells. Chronic inflammation results in progressive fibrosis and thickening of the adventitia and intima, resulting in increased vessel wall thickness and plaque formation. 19 When large or circumferential, these plaques cause narrowing of the aortic lumen in a ‘skip’ pattern with interspersed areas of healthy aorta in a pattern that is characteristic of TA.
By contrast, GCA is characterised by more prominent inflammation of the inner media and intima, with the adventitia being less commonly affected. 16 These acellular areas act as a focus of calcification, which in turn trigger a ‘foreign body-like’ inflammatory reaction in which granulomas form around areas of atrophic calcified medial tissue.20,21
Genetic associations in TA and GCA
A large international multicentre seminal study published in 2013 which genotyped ∼200,000 genetic variants in two ethnically divergent TA cohorts from Turkey and North America (n = 451) identified two independent susceptibility loci within the human leukocyte antigen (HLA) region HLA-B/MICA and HLA-DQB1/HLA-DRB1, as well as a genetic association between IL12B and TA. 22 More recent genome-wide association studies (GWAS) have also identified associations between TA and several non-HLA regions. Interestingly, the allele identified has implications in terms of disease burden. 23 Kasuya et al. 24 and Sahin et al. 25 have both published findings that the severity of disease is worse in those with HLA-B*52. In contrast, Kitamura et al. 26 published the results of 85 patients with TA identified with HLA-B39 (B*3901 and B*3902) alleles who had significantly increased frequency of renal artery stenosis. Conversely, small prospective cohort studies have found several HLA-B alleles (B*6701, B*5201, and B*4006) to be protective from TA, but have not reached the threshold for genome-wide significance and require further clarification. 27
Besides inflammation being a key histopathological feature, substantial evidence supports the hypothesis that GCA is an autoantigen-driven disease. Genetic studies have demonstrated that GCA is associated with the major histocompatibility complex (MHC) class II molecule HLA-DRb1*04.28–30 Interestingly, small studies have described associations with gene polymorphisms for intercellular adhesion molecule (ICAM)-1, CCL5 (RANTES, regulated upon activation, normal T-cell expressed, and presumably secreted), and interleukin (IL) 1 receptor. 31
Shared immunopathogenesis of TA and GCA
One of the strongest arguments for TA and GCA sharing the same pathological process arises from the similarities in the inflammatory infiltrate present in the aortic wall (Figures 2 and 3).
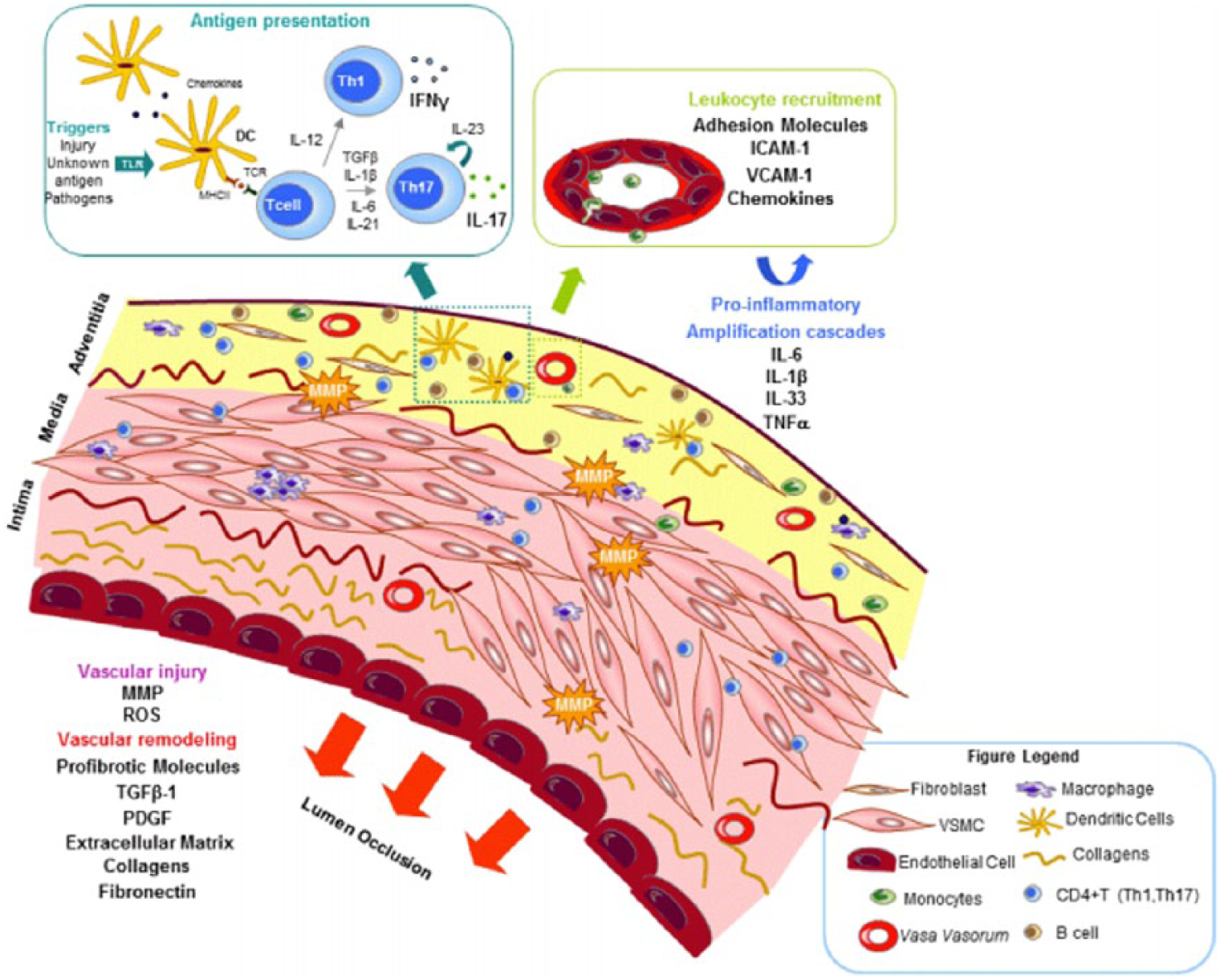
Summary representation of immunopathogenic mechanisms involved in vascular inflammation and remodelling in giant cell arteritis. Reproduced with permission from OMICS International. 99
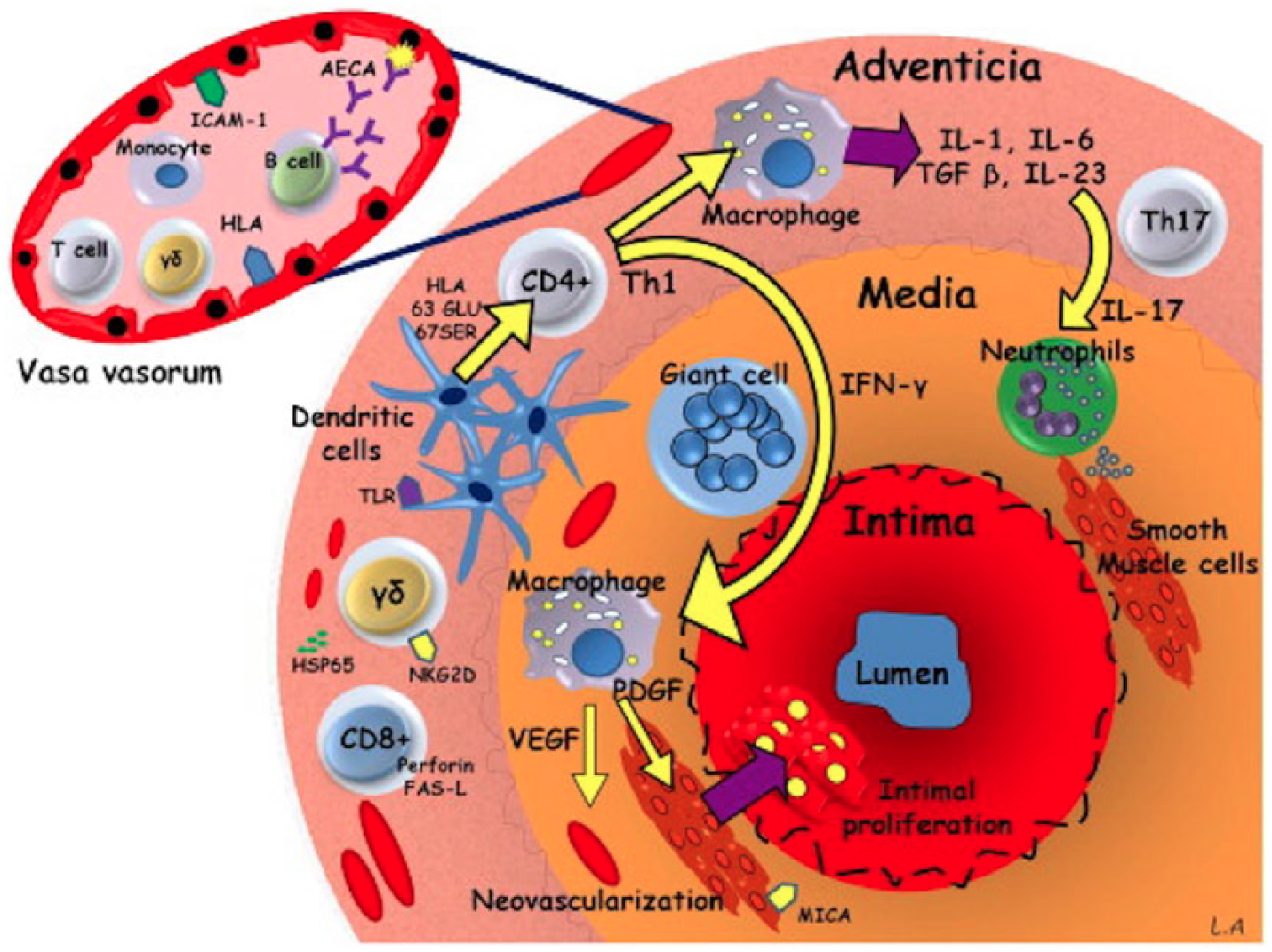
Summary representation of immunopathogenic mechanisms involved in vascular inflammation and remodelling in Takayasu arteritis. Reproduced with permission from Elsevier. 67
Innate immunity in large vessel vasculitis (LVV)
As both the media and intima are avascular, the site of primary injury is thought to be the vasa vasorum within the adventitia. In LVV, dendritic cells are activated by Toll-like receptors (TLR) via an unknown trigger. Weyland et al. 32 demonstrated the crucial role of dendritic cells (DCs) in LVV using a mouse model of GCA in which temporal artery biopsies from GCA patients were grafted into mice with severe combined immunodeficiency (SCID). Using this model, the presence of anti-DC antibodies were seen to protect against the development of inflammation. 33 Unlike in physiological immune response, vascular wall DCs in GCA fail to migrate to the local lymph node, where they produce the chemokines CCL19 and CCL21 while simultaneously expressing their ligand CCR7, resulting in them becoming trapped in the arterial wall where they trigger local immunity. 34 Trapped DCs become activated and recruit CD4+ T-cells via the differential expression of TLRs, which can dictate the localisation and extension of the inflammatory response. Specifically, TLR4 ligands trigger the recruitment of CD4+ T-cells that infiltrate all layers of the arterial wall inducing a transmural panarteritis, whereas TLR5 ligands trigger an adventitial perivascular infiltrate; therefore, TA and GCA are associated with specific TLRs.35,36
Interestingly, DCs have also been implicated in controlling which vessels are affected in LVV as different arteries are known to express different combinations of TLRs 1–9. Pryshchep et al. 36 showed that this differential expression results in vessel-specific T-cell activation profiles and concluded that this may account for the variable involvement of arteries by different inflammatory vasculopathies. Once activated, mature DCs express CCL20, which binds to its ligand CCR6 on the surface of circulating CD4+ T-cells, resulting in their recruitment. DCs then trigger the adaptive immune response by presenting an as yet unknown antigen to CD4+ T-cells within the arterial wall.
Finally, neutrophils appear to play a role in the pathogenesis of LVV but this role is poorly understood and has rarely been studied. A role for neutrophils is suggested by the high levels of circulating IL17 in LVV, which is known to mobilise neutrophils from bone marrow as well as trigger their activation and recruitment to perivascular tissue. 37
Adaptive immunity in LVV
Once the immune system has been triggered and the cascade has been activated, CD4+ T-cells infiltrate the media, proliferate and secrete a range of pro-inflammatory cytokines, resulting in further T-cell differentiation. Pro-inflammatory cytokines stimulate the further differentiation of CD4+ T-cells to either T-helper (TH) 1 (IL12 or IL18) or TH17 (IL6, IL1β or IL23) cells.38,39 Nevertheless, there are differences between the pathogenesis of TA and GCA in terms of the T-cell subsets that they display and their distinct cytokine signatures. In GCA, the TH17 cell pathway, and its accompanying cytokines, were found to be suppressed by glucocorticoid treatment, while the TH1 pathway was resistant. 40 Interestingly, Saadoun et al. report the opposite finding in TA, 39 with only the TH1 pathway suppressed by steroids.
In recent years, a subset of T-lymphocytes producing IL9 (TH9 cells) has been proposed to play a role in the pathogenesis of LVV. Pan et al. demonstrated increased IL9 levels in the serum of 11 patients with active TA together with significantly increased numbers of circulating TH9 lymphocytes. 41 Increased TH9 cells and IL9 levels have also been found in GCA patients displaying a characteristic pattern of transmural inflammation and small vessel vasculitis. 42
The final phase in the pathogenesis of both TA and GCA involves remodelling of the vessel wall resulting in vessel stenosis, occlusion or aneurysm. In GCA, interferon gamma (IFNγ)-activated macrophages produce a number of cytokines that amplify the inflammatory response (IL6, IL1β, and tumour necrosis factor alpha (TNFα)) as well as mediators of vascular remodelling such as matrix metalloproteinases (MMPs), which act to degrade components of the extracellular matrix.43,44 Nikkari et al. demonstrated that MMP-9 containing macrophages colocalised with sites of internal elastic lamina degradation in GCA. 45 Similarly, temporal artery biopsies of 147 GCA patients showed upregulated MMP-9 expression that was associated with intimal hyperplasia, luminal narrowing and neoangiogenesis. 46 A probable role of MMPs in the pathogenesis of TA is also supported by the finding of significantly increased expression of MMP 1, 3, and 9 in patients with active disease. 47 Further evidence of the role of MMPs in the pathogenesis of TA is provided by the positive correlation between oxidative stress and levels of MMP, 48 and a negative association of the antiatherogenic molecule sRAGE with MMP. 49 As such, MMPs are being investigated as a potential therapeutic target for TA and as biomarkers of disease activity. 50 Activated macrophages, giant cells, and injured vascular smooth muscle cells (VSMCs) also release vascular endothelial growth factor (VEGF) and platelet-derived growth factor (PDGF), resulting in neovascularisation and intimal proliferation hyperplasia.51,52
Potential pathogenic triggers of LVV
Both infective agents and autoimmune mechanisms have been proposed as potential triggers of inflammation in LVV. Evidence suggesting a role of mycobacterial proteins as a trigger in TA includes an increased humoral immune response directed towards the mycobacterial 65 kDa heat-shock protein (mHSP65) in TA patients,53–56 and the finding that a 65-kDa HSP is induced in the vasa vasorum and media of temporal artery biopsies. 53 However, clinical studies have shown that only a small minority of patients with TA have a history of exposure to mycobacteria. 57 Furthermore, Arnaud et al. failed to find any evidence for the presence of mycobacteria in arterial samples from 10 TA patients using acid-fast staining, mycobacterial culture and direct amplification of the Mycobacterium tuberculosis genome. 58
Several pathogens, primarily viruses, have also been investigated as potential triggers of GCA. No consistent association was found between the incidence of GCA and human herpes virus (HHV)-6, HHV-7, Epstein–Barr virus, cytomegalovirus, herpes simplex virus and human parvovirus B19.59,60 Recently, Gilden et al. found a significant association with varicella zoster virus (VZV) and GCA.61,62 However, recent studies by other groups have not corroborated the findings that reactivation of VZV might trigger the immune response in GCA.63,64 Finally, Wagner et al. demonstrated an association between Chlamydia pneumoniae DNA and GCA in six patients. 65 However, five subsequent studies failed to corroborate this finding. 66 To date, no consistent evidence exists for a microbiological cause for LVV, although opinion is divided on the potential role of VZV as a trigger for GCA.
A number of studies have reported detection of various autoantibodies in TA patients but it is not known whether these directly contribute to the pathogenesis of TA or represent by-products of immune activation. 67 Immune complexes and antiaortic antibodies have been reported in several studies.68,69 However, the aortic autoantigen recognised by autoantibodies remains unknown. Dhingra et al. 70 showed that collagenase-digested aortic tissue induced a fall in the antibody titre of aortitis patients, suggesting collagen as a candidate autoantigen. However, when Baltazares et al. attempted to find a triggering antigen within components of the aortic wall, such as elastin, fibronectin, and collagen, no differences were found between TA patients and controls in identifying a selective triggering factor separate from each other. 71 Similarly, studies performed in GCA have also failed to find consistent evidence of a triggering antigen. Schmits et al. screened a cDNA library derived from human testes for antigens reacting with sera of GCA patients and identified only two antigens that reacted specifically in 32% of GCA cases: lamin C and the nuclear antigen of 14 kDa. 72
The presence of antiendothelial cell antibodies (AECAs) has been demonstrated in both TA and GCA and has been suggested as possible triggers of inflammation. There is some evidence in TA that AECAs can induce endothelial cell apoptosis via complement-mediated pathways.73,74 A pathogenic role of AECAs in TA is supported by the findings of Chauhan et al. 75 who showed that the sera of TA patients can only induce endothelial cell death in the presence of AECAs and Wang et al. 76 who reported increased levels of AECAs from circulating B-lymphocytes in TA compared to normal controls. AECA levels were also found to be higher in TA patients with active disease compared to inactive disease. 76 However, the presence of increased AECA in disease is not sufficient to imply causation, as their production may be induced in response to endothelial cell death itself. 77 AECAs have also been demonstrated in 33% of patients with GCA. 78 However, it should be noted that AECAs have also been demonstrated in healthy controls as well as numerous systemic autoimmune disease.79,80 As such, their presence is not disease-specific and no studies have yet demonstrated a pathological role for AECAs in GCA.
Biomarkers – the search continues
C-reactive protein (CRP) and erythrocyte sedimentation rate (ESR) remain the mainstays for monitoring disease activity despite neither being shown to be sensitive or specific enough for the task. 81 Despite this, in clinical practice, no other biomarkers have been developed for meaningful clinical correlation.
Serum levels of IL6 have been found to be more sensitive than ESR in indicating disease activity, 82 while VEGF levels are upregulated after recent episodes of optic nerve ischaemia, 83 and down-regulated in response to treatment. 84 Sun et al. demonstrated that levels of MMP-9 correlated strongly with severity of vessel lumen stenosis and wall thickness on cross-sectional imaging of TA patients, with stronger associations than either CRP or ESR. 85 Finally, Springer et al. have investigated patients with both GCA and TA and found that serum levels of different S100 proteins (S100A8/S100A9 and S100A12) in GCA were significantly elevated during active disease and although there were weak correlations between levels of S100 proteins and ESR or CRP, a model including S100A8/S100A9, S100A12, ESR, and CRP was a better indicator of disease activity compared to ESR and CRP together. S100, however, did not show any significant differences in active or inactive TA. 86
Advances in medical therapy for TA and GCA
Currently the medical treatment of choice for both TA and GCA is systemic administration of corticosteroids aimed at dampening the immune response. Whilst this is an effective treatment, there are considerable side effects associated with long-term steroid use. In a study of 125 patients with GCA, Proven et al. demonstrated an 86% risk of developing steroid-associated complications including bone fracture, avascular necrosis, diabetes mellitus, and hypertension. 87 Furthermore, steroid therapy has been shown to have little effect in preventing relapses. 88 Corticosteroid therapy is often supplemented with the addition of a further immunosuppressive agent such as azathioprine, methotrexate, cyclophosphamide, cyclosporin, rapamycin or mycophenolate mofetil. In general, the results of treatment trials with these conventional immunosuppressive agents have been disappointing. 89
Ferfar et al. reviewed 13 studies including 96 TA patients treated with anti-TNFα. The median daily prednisolone dose pre- and post-anti-TNFα treatment was 15 mg/day and 8 mg/day, respectively. There was also a reduction in median CRP from 30 mg/L to 9 mg/L, and 61.4% of treated patients reported improvement in clinical symptoms. 90 As such, anti-TNFα treatment appears to have some efficacy in reducing the burden of inflammation in TA, and allows reduction of maintenance steroid regimes. A review of the literature suggests that no randomised controlled trials (RCTs) of anti-TNFα agents in the treatment of TA have been performed. The rarity of TA makes undertaking such trials extremely difficult even with multicentre and international collaboration. This is highlighted by the finding that one of the largest series to date of anti-TNFα treatment is by Mekinian et al., which contains 35 patients and is a multicentre retrospective study over a 12-year period. 91
Langford et al. have published results of a multicentre randomised double-blind trial, a rarity in TA, investigating the effect of abatacept (CTLA-4Ig) in treating TA. 92 Of the 34 patients enrolled, all initially received intravenous abatacept with prednisolone for 12 weeks and then proceeded (if in remission) to randomisation to receive either placebo or continued abatacept. The relapse-free survival rate did not reach significance at 12 months (22% vs 40% for abatacept vs placebo, respectively (p = 0.853)). Although the trial did not show any difference between abatacept or placebo, it should certainly be noted that of the 34 patients initially recruited only 26 went on to be randomised.
Seror et al. showed no improvement in remission rates when adalimumab was added to standard GC treatment in a RCT of 70 patients. 93 A further randomised trial of infliximab versus placebo in addition to GC failed to show any improvement in remission maintenance in 44 patients and the use of etanercept versus placebo also showed no additional benefit in an RCT of 17 patients.94,95 After 12 months, 50% of patients treated with etanercept remained in remission compared to 22% of the placebo group (p = NS).
Several studies describe the use of tocilizumab (TCZ) in GCA. Until recently, these studies predominantly consist of small case series and individual case reports. A single-centre RCT randomised 20 patients to TCZ and 10 to placebo in addition to standard steroid therapy. Complete remission was found in 85% of the TCZ group compared to 40% of the placebo group at 12 weeks (p = 0.03). One-year recurrence-free survival was achieved in 17 patients treated with TCZ and two patients treated with placebo. 96 Results from the much anticipated GiACTA trial, a multicentre RCT which included 251 patients, has recently published its results comparing different doses of TCZ or placebo with tapering prednisolone. 97 Following 52 weeks of follow-up, patients receiving TCZ, regardless of dose, showed a significantly reduced rate of sustained glucocorticoid-free remission (56% and 53% treated with TCZ weekly or every other week, respectively, compared with 14% and 18% in the placebo group that underwent the 26-week prednisone or 52-week prednisone taper (p < 0.001 for the comparisons of either active treatment with placebo)).
Langford et al. performed a multicentre double-blind RCT of abatacept use in GCA. 98 Abatacept (CTLA-4Ig) prevents antigen presenting cells such as DCs from providing costimulatory signals to T-cells, thus blocking their activation. Langford et al. recruited 49 patients with GCA who underwent treatment with both prednisolone and abatacept for 12 weeks. At this time, those in clinical remission (n = 41) underwent double-blind randomisation to abatacept or placebo. After standardised steroid weaning, 48% of patients on abatacept were found to have remained in remission at 12 months compared to 31% of the placebo group (p = 0.049). The authors conclude that the addition of abatacept to a GCA treatment regimen with prednisolone reduces the risk of relapse and is not associated with a higher rate of toxicity compared to prednisolone alone. The evidence for use of TCZ in GCA is supported by two RCTs, with the results of the larger GiACTA trial showing very promising results. However, the benefit of anti-TNFα agents has not yet been shown, although well-powered studies are lacking. Despite evidence of the role of MMPs and their regulatory tissue inhibitors of metalloproteinases (TIMPs) in the pathogenesis of collagen and elastin degradation in LVV, no therapeutic agents have yet been developed against these targets. However, they remain a potential future target for novel therapies.
Future challenges and controversies
Over the last decade, increased understanding of the immunopathogenesis of TA and GCA has led to the identification of new exciting avenues for future advancements both in terms of diagnostic biomarker development and medical therapy. However, the triggering pathological agent underlying these conditions remains elusive and its identification represents the most significant future challenge in the management of LVV. The identification of the causative agents in TA and GCA will likely represent the only reliable method of clarifying the ongoing question as to whether these conditions are distinct or subtypes of a single disease entity. In clinical practice, such a distinction is largely irrelevant unless it informs management and improves patient outcomes; hence, recognition of different types of TA and GCA is beneficial only if the treatment priorities change, such as using steroids as an urgent treatment line in cases of TA patients. The main advantage in identifying the triggering stimulus lies in enabling diagnosis and treatment earlier in the disease process and minimising destruction of the elastic walls of large vessels. If an infective trigger is found to be responsible then curative treatments can be targeted.
Conclusions
Controversy still surrounds the classification of TA and GCA as separate disease entities. Clinical presentation, age of onsets and racial distribution are distinct between the two diseases. Furthermore, although histological analysis can demonstrate considerable overlap between the conditions, specific histopathological features characterise each condition. Similarly, recent advances in the genetic profiling of the two conditions have shown that, as yet, there is no evidence to suggest a common genetic background in LVV, with TA and GCA being primarily associated with different HLA loci. However, in recent years, a common immunopathological basis of LVV has emerged consisting of activation of adventitial dendritic cells, recruitment of CD4+ T-lymphocytes and infiltration of macrophages, resulting in cytokine driven neovascularisation and intimal hyperplasia. As such, it is still unclear as to whether these two conditions truly are separate disease entities. Crucial to resolving this matter is the elucidation of the underlying cause of vessel wall inflammation, with both infective agents and autoantigens being investigated as potential triggers. A better understanding of the underlying aetiology of LVV would also aid in the development of early diagnostic and treatment modalities aimed at preventing the dramatic consequences of aortic luminal narrowing, aneurysm, and dissection.
Footnotes
Declaration of conflicting interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors received no financial support for the research, authorship, and/or publication of this article.