
Abstract
Increasing use of the new oral anticoagulants (NOACs) – dabigatran, rivaroxaban, and apixaban – has prompted considerable discussion in the medical community even as warfarin remains the mainstay of therapy. This article raises 10 controversial issues regarding the use of NOACs for stroke prevention in patients with atrial fibrillation, and offers a review of the latest available evidence. We provide a brief overview of the mechanism and dosing of these drugs, as well as a summary of the key clinical trials that have brought them into the spotlight. Comparative considerations relative to warfarin such as NOAC safety, efficacy, bleeding risk, reversibility, drug-transitioning and use in patients well controlled on warfarin are addressed. Use in select populations such as the elderly, those with coronary disease, renal impairment, or on multiple anti-platelet drugs is also discussed. Finally, we consider such specific issues as comparative efficacy, off-label use, cost, rebound and management during events. Ultimately, the rise of the NOACs to mainstream use will depend on further data and clinical experience amongst the medical community.
Introduction
Since its approval for therapeutic use in 1954, warfarin has remained the mainstay of oral anticoagulant therapy across multiple clinical indications. Despite aggressive attempts to develop agents that would be safer, more predictable and better tolerated, it has only been within the last few years that viable alternatives have arrived, three of which have been approved for clinical use in the US for patients with non-valvular atrial fibrillation (NVAF). One of these is also approved for venous thromboembolism (VTE) prophylaxis after knee or hip replacement surgery. In order of completion of large phase III clinical trials, dabigatran, rivaroxaban, apixaban and edoxaban (not FDA approved) – commonly termed the new oral anticoagulants (NOACs) – have been met with great anticipation by the medical community. Advantages of these agents over warfarin include fixed-dosing, once or twice daily (BID) regimens, rapid and predictable onset of anticoagulant effect, a wider therapeutic window between antithrombotic efficacy and the bleeding risk, few food or drug interactions, and no necessity of routine anticoagulant intensity monitoring. 1 Faced with an aging population, the rising prevalence of atrial fibrillation (AF) (an estimated 17.8% among those over age 85 years), and the aggregate 64% reduction in stroke risk conferred by warfarin compared to placebo or no therapy, the potential healthcare and economic impact of the NOACs is unquestionable.2,3
Although the development of these drugs is a landmark event in cardiovascular therapeutics, an array of issues has dampened the initial enthusiasm, and warfarin remains the most commonly prescribed anticoagulant even for patients newly starting therapy for NVAF. Clinicians must reconcile the benefits of NOACs with their drawbacks, including lack of an antidote or methods to assess compliance, cost, uncertainties in patients with kidney disease, and a tendency to migrate toward off-label use. However, scrutiny surrounding these issues has invigorated the discussion surrounding NOACs and promises not only to enhance understanding of these drugs and the mechanisms underlying thromboembolic events, but ultimately to improve patient outcomes. Following is a brief review of the pivotal, phase III clinical trials of the four NOACs and a discussion of the decisions that underlie their safe and appropriate use.
Mechanism and dosing
While vitamin K antagonists inhibit the production of several factors in the coagulation cascade, newer agents target single steps, resulting in greater predictability of anticoagulant effect (Figure 1). 4 Dabigatran is a direct thrombin inhibitor (DTI) that impairs the ability of thrombin to (i) convert fibrinogen to fibrin, (ii) promote self-propagation by activation of other coagulation factors, and (iii) activate platelets. 5 Unlike indirect inhibitors of thrombin such as heparin, dabigatran reversibly binds the catalytic site of both free and clot-bound thrombin. 6 Administered as an inactive prodrug, dabigatran undergoes complete esterase-mediated conversion to its active form, reaches peak plasma levels within 2–3 hours and has a half-life of 12–17 hours. 7 The usual dose is 150 mg BID. A lower dose of 110 mg BID, approved in other parts of the world for patients over age 75 or at high risk of bleeding, is not available in the US. The FDA approved 75 mg BID for patients with creatinine clearance (CrCl) of 15–30 ml/min based on pharmacokinetic models predicting that this would result in plasma concentrations of the drug comparable to standard dosing in patients with normal renal function. 8
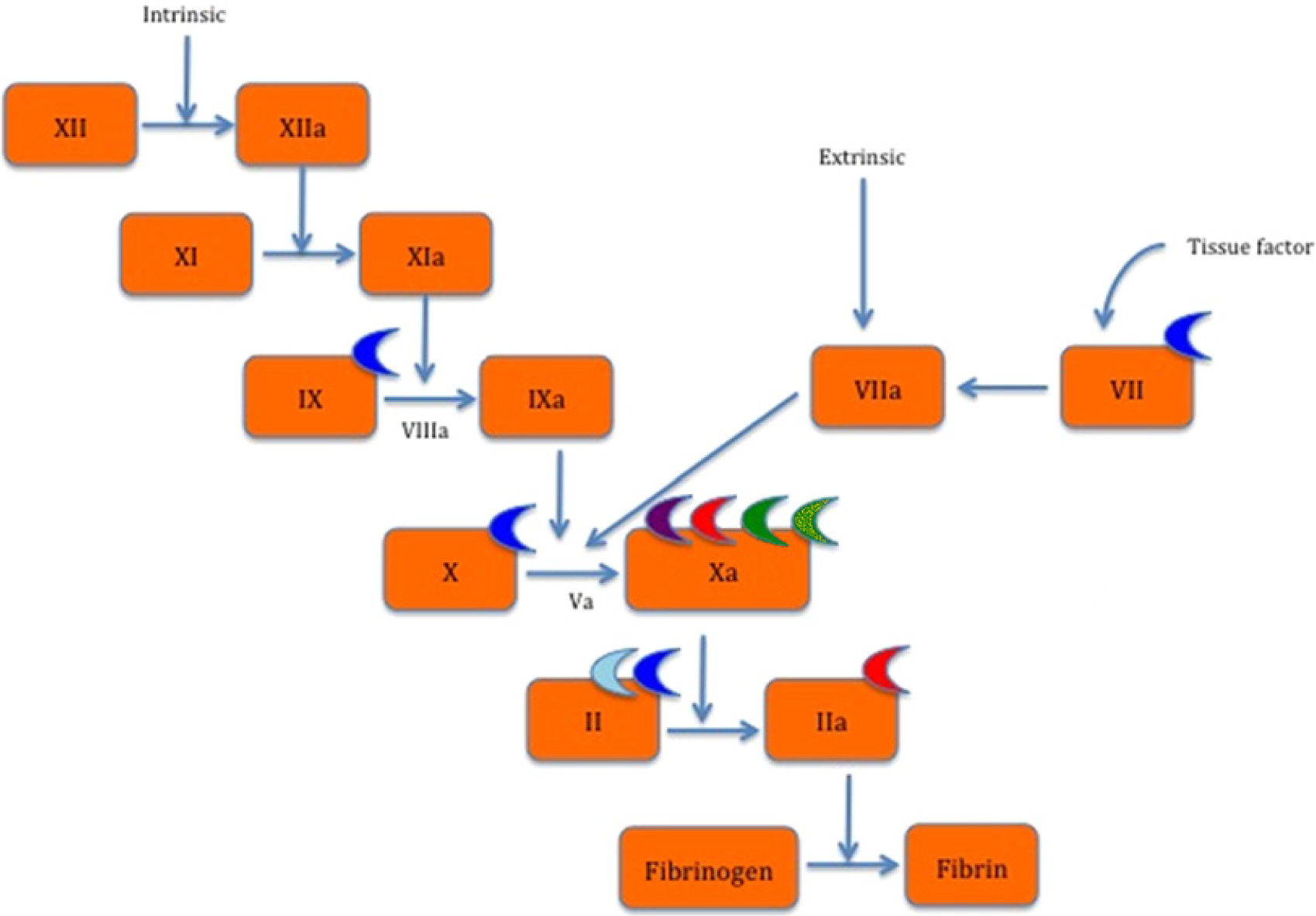
Anticoagulant site of action. 4
Rivaroxaban, apixaban and edoxaban are extremely selective, reversible, direct factor Xa inhibitors. Rivaroxaban reaches peak plasma levels 2–4 hours after administration, is dosed at 20 mg once daily with a half-life of 5–13 hours, and should be taken with food to increase bioavailability. 9 A 15 mg dose is available for patients with a CrCl of 15–30 ml/min, as 33% of absorbed rivaroxaban is excreted unchanged renally, while the remainder is metabolized by both cytochrome P450-dependent and independent processes, including CYP3A4, CYPP2J2, and P-glycoprotein. 1 Caution should be exerted when using rivaroxaban in conjunction with pharmacologic inducers or inhibitors of these enzymes. A dose of 10 mg once daily is used for prophylaxis of VTE following major orthopedic surgery of the hip or knee. Twice daily dosing is indicated for acute coronary syndromes. It is also approved for treatment of venous thromboembolism.
Apixaban reaches peak plasma levels 3–4 hours after administration, has a half-life of 8–15 hours and is dosed at 5 mg BID. 10 Approximately a quarter of the administered dose is excreted by the kidneys unchanged and the remainder is metabolized by CYP3A4-dependent and independent mechanisms, and should be avoided with other drugs metabolized in the same manner. A 2.5 mg BID dose is available for patients with two or more of the following criteria: age greater than 80 years, body weight less than 60 kg or serum creatinine above 1.5 mg/dl. 1
Edoxaban is rapidly absorbed, reaching peak plasma levels 1–2 hours after oral administration and has a half-life of 9–11 hours.11,12 It has a bioavailability of 50%, and there is no inactive pro-drug requiring modification into an active form. The drug is eliminated via two mechanisms. Approximately 35% undergoes renal elimination, while the remainder is excreted in the feces. 13 Dosing regimens have not yet been defined for widespread use, but in the ENGAGE AF-TIMI 48 (Effective Anticoagulation with Factors Xa Next Generation in Atrial Fibrillation – Thrombolysis in Myocardial Infarction 48) trial, patients received either 60 mg or 30 mg fixed daily dosing, with rates of stroke or systemic embolism comparable to those with warfarin and significantly lower rates of bleeding and death from cardiovascular causes. 14 Edoxaban is also a potent inhibitor of cytochrome P450 3A4 and P-glycoprotein 13 (Table 1).
Characteristics of the anticoagulant drugs dabigatran, rivaroxaban, apixaban, edoxaban and warfarin.
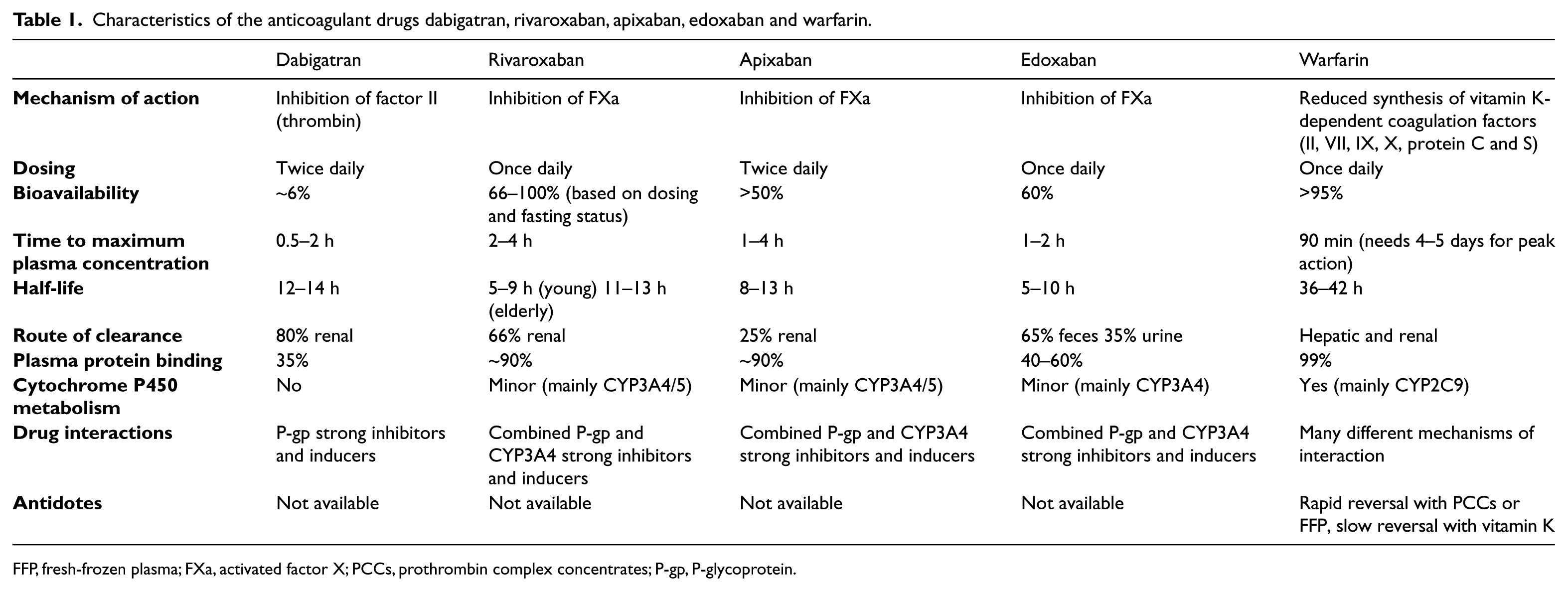
FFP, fresh-frozen plasma; FXa, activated factor X; PCCs, prothrombin complex concentrates; P-gp, P-glycoprotein.
Pivotal trials in patients with NVAF
Dabigatran
Approval of dabigatran was based on the RE-LY (Randomized Evaluation of Long term anticoagulant therapY) trial, a phase III study that randomized 18,113 patients with NVAF and at least one additional risk factor for stroke (mean CHADS2 score = 2.1) to one of two blinded doses of dabigatran (110 mg BID or 150 mg BID) or open-label, adjusted-dose warfarin (goal INR 2.0–3.0). 15 For the primary outcome of all stroke (ischemic or hemorrhagic) and systemic embolism, the rate was 1.7%/year with warfarin, 1.5%/year with dabigatran, 110 mg BID (non-inferior: HR=0.90, 95% CI 0.74–1.10, p<0.001) and 1.1%/year with 150 mg BID (superior: HR=0.65, 95% CI 0.52–0.81, p<0.001). Major bleeding was significantly lower with dabigatran 110 mg BID compared to warfarin (3.6% to 2.9%/year, p=0.003), whereas there was no difference in major bleeding in those treated with dabigatran 150 mg BID and warfarin (3.3%, p=0.31). Rates of intracranial bleeding and hemorrhagic stroke were significantly lower in both dabigatran groups than with warfarin (p<0.001), but gastrointestinal (GI) bleeding was more frequent in the 150 mg BID dabigatran group (p<0.001) compared to warfarin. Total mortality was not significantly different between the three groups over a median of 2 years. A trend toward higher rates of myocardial infarction with either dose of dabigatran compared to warfarin did not reach statistical significance when silent myocardial infarction (MI) events were included in the analysis. 16
At study centers where the mean time in therapeutic range (TTR) on warfarin was lower than the trial mean of 64%, either dose of dabigatran was associated with lower rates of mortality, major bleeding, and composite ischemic and bleeding outcomes, though there was no difference in rates of the primary outcome, suggesting that dabigatran may benefit patients who are poorly controlled with warfarin. Notably, patients with CrCl <30 ml/min were excluded from the trial, and half the patients were naïve (<60 days of therapy) to warfarin at entry.
Initially after regulatory approval, a number of bleeding events were reported to the FDA, prompting further investigation. Using insurance-claims data and administrative data from the FDA Mini-Sentinel database, a pilot program of the Sentinel Initiative, reviewers found bleeding rates with dabigatran similar to those of warfarin – commensurate with the results of the RE-LY trial. Ongoing post-market analyses, including review of the Mini-Sentinel database and others that adjust for patient-level variables, are underway. 17
Rivaroxaban
The ROCKET AF (Rivaroxaban Once Daily Oral Direct Factor Xa Inhibition Compared with Vitamin K Antagonism for Prevention of Stroke and Embolism Trial in Atrial Fibrillation) study was a double-blind study in which 14,264 patients with NVAF and CHADS2 scores ≥2 (mean 3.5) were randomized to rivaroxaban 20 mg once daily or dose-adjusted warfarin (goal INR 2.0–3.0); patients with CrCl 30–49 ml/min received rivaroxaban at 15 mg daily, and those with CrCl below 30 ml/min were excluded. 18 There were 37.6% of warfarin-naïve patients. After a median follow-up of 1.93 years, rates of the primary outcome of stroke or systemic embolism were 2.1% per 100 patient-years in the rivaroxaban arm and 2.4% with warfarin (HR=0.88, 95% CI 0.74–1.03, p<0.001 for non-inferiority), though the reduction in ischemic stroke was not significant. Among these older, sicker patients, the mean TTR on warfarin was lower than in other trials at 55%. The annual rate of major bleeding was not different between treatment groups, though those assigned to rivaroxaban had a lower rate of intracranial bleeding (0.5%/year vs 0.7%/year, p=0.02) and, as a result, less fatal bleeding (0.2%/year vs 0.5%/year, p=0.003). Total mortality was not significantly different between groups. A trend toward lower rates of myocardial infarction in the rivaroxaban group did not reach statistical significance. Gastrointestinal bleeding and transfusion requirements were greater with rivaroxaban, particularly in elderly male patients with prior GI bleeding. This was further described in a post hoc analysis which showed that non-major (1.75% vs 1.39%/year, HR=1.26, 95% CI 1.20–1.55) and major bleeding (2.0% vs 1.24%/year, HR=1.61, 95% CI 1.30–1.99) were greater with rivaroxaban, but fatal bleeding events were less frequent than with warfarin. 19
Apixaban
Apixaban more recently received FDA approval based on the AVERROES (Apixaban VERsus acetylsalicylic acid to pRevent strOkE in atrial fibrillation patientS who have failed or are unsuitable for vitamin K antagonist treatment) and ARISTOTLE (Apixaban for Reduction of Stroke and Other Thromboembolic Events in Atrial Fibrillation) trials. 20 In AVERROES, 5599 patients with NVAF and at least one additional stroke risk factor either unwilling or deemed ineligible to take vitamin-K antagonists (VKAs) were randomized in double-blind fashion to apixaban 5 mg BID, or aspirin 81–324 mg daily. 21 A reduced dose of 2.5 mg BID was given to patients with two or more of the following criteria: age ≥80 years, weight ≤60 kg, or serum creatinine ≥1.5 mg/dl. The study was terminated by recommendation of the Data and Safety Monitoring Board (DSMB) after 1.1 years’ median follow-up because of a marked benefit of apixaban. The primary outcome of stroke and systemic embolism was reduced from 3.7%/year with aspirin to 1.6%/year with apixaban (HR=0.45, 95% CI 0.32–0.62, p<0.001). There was no difference in rates of major bleeding or intracranial bleeding. At 2 years, rates of study drug discontinuation were higher in the aspirin group than in the apixaban group (20.5%/year vs 17.9%/year, p=0.03).
ARISTOTLE was a double-blind phase III study randomizing 18,201 patients with NVAF and one or more additional stroke risk factors to apixaban using the same dosing regimen as in AVERROES or dose-adjusted warfarin (goal INR 2.0–3.0). 22 Patients with CrCl <25 ml/min were excluded. Forty-three per cent of the patients were warfarin-naïve (maximum 30 days’ warfarin use prior to randomization). The mean CHADS2 score was 2.1, similar to the RE-LY population, but less than the ROCKET AF population; median treatment duration was 1.8 years. In the apixaban group there was a significant reduction in stroke or systemic embolism compared to warfarin (1.6% to 1.3%/year, HR=0.79, 95% CI 0.66–0.95%/year, p=0.01) but no significant difference in rates of ischemic stroke. Major bleeding was reduced in the apixaban group (3.1% to 2.1%/year, p<0.001), reflecting reduced rates of both hemorrhagic stroke and intracranial bleeding. Apixaban was more effective in reducing the risk of intracranial bleeding than warfarin in patients at higher risk of bleeding stratified by HAS-BLED scores. 23 There was no difference in GI bleeding between groups. Apixaban demonstrated a marginally significant reduction in mortality compared to warfarin from 3.9%/year to 3.5%/year (p=0.047). A trend toward lower rates of MI was not statistically significant. Fewer patients in the apixaban group discontinued therapy compared to warfarin (25.3% vs 27.5%). A pre-specified secondary analysis found no significant interaction between TTR (mean of 62%) in the warfarin group and safety or efficacy outcomes. A subsequent analysis also showed that both safety and efficacy outcomes were maintained across all levels of individual TTR and quality of center-specific INR control, with the greatest effect in centers with low-quality control and in patients with low predicted TTR, but still significant in patients and centers with higher-quality control. 24
Edoxaban
The ENGAGE AF-TIMI 48 (Effective Anticoagulation with Factors Xa Next Generation in Atrial Fibrillation – Thrombolysis in Myocardial Infarction) study was a randomized, double-blind, double dummy placebo-controlled trial involving 21,105 patients with atrial fibrillation at moderate to high risk of stroke (CHADS2 scores ≥2) randomized to edoxaban, 30 or 60 mg daily or dose-adjusted warfarin in a 1:1:1 fashion. 14 The dose of edoxaban was halved in patients with CrCl 30–50 ml/min, body weight <60 kg or concomitant treatment with quinidine, verapamil or dronedarone.
After a median follow-up of 2.8 years, in the modified intention-to-treat analysis, the annualized rate of the primary endpoint of stroke or systemic embolism was 1.50% with warfarin versus 1.18% with high-dose edoxaban (HR=0.79, 97.5% CI 0.63–0.99, p<0.001 for non-inferiority) and 1.60% with low-dose edoxaban (HR=1.07, 97.5% CI 0.87–1.31, p=0.005 for non-inferiority). The risk reduction was driven mainly by lower rates of hemorrhagic stroke. The mean TTR on warfarin was higher than in other trials at 68.4%. Subgroup analysis revealed that VKA-naïve patients had significantly fewer strokes or systemic embolic events with high-dose edoxaban than with warfarin, and that aspirin and amiodarone use in patients randomized to the low dose of edoxaban improved the treatment effect.3,25
Rates of major bleeding were lower in both groups compared to warfarin, though fatal bleeding rates was similar. Compared to warfarin, edoxaban was associated with much lower rates of intracranial bleeding: 0.85% versus 0.39% of patients/year (HR=0.47, 95% CI 0.34–0.63, p<0.001, NNT=217). There was a statistically significant improvement in cardiovascular mortality in both the high-dose (HR=0.86, 95% CI 0.77–0.97, p=0.013) and low-dose group (HR=0.85, 95% CI 0.76–0.96, p=0.008). Gastrointestinal bleeding was more frequent in the high=dose edoxaban group than with warfarin but was less in the low-dose edoxaban group.
The ‘well-controlled’ warfarin patient
Given the established practice patterns and systems to manage patients on warfarin therapy and the uncertainty surrounding the NOACs, clinicians might find comfort in continuing patients on ‘well-controlled’ warfarin rather than initiating a new agent. Identifying these patients is challenging because few patients are consistently therapeutic. In a study of VA patients, the mean TTR was 66.5% and varied widely among patients. 26 The lack of a uniform definition further makes these patients very difficult to quantitatively identify. Moreover, a population-based study of more than 125,000 individuals on warfarin for AF – 69% with CHADS2 scores ≥2 – found that more than 60% stopped therapy by 5 years, especially younger males with lower CHADS2 scores. 27 The median time to discontinuation was 2.9 years. This suggests that the term ‘well-controlled’ may be more an exception among warfarin-treated patients than the norm. Proponents of NOACs argue that the term ‘well controlled’ offers a false sense of security, nested primarily in a need for frequent INR monitoring rather than comparative clinical efficacy and safety. An analysis of all outcomes in RE-LY by quartiles of TTR showed no interaction with outcomes in either dose group, though the advantage of dabigatran was greatest at sites with poor INR control. 28 Superiority of the 150 mg BID dose, non-inferiority of the 110 mg BID dose, and reduced intracranial bleeding with either dose were maintained across all quartiles of TTR. Though the validity of such subgroup stratification is questionable, a similar analysis of ROCKET AF revealed no significant interaction between a center’s mean TTR and outcomes, and no quartile in which warfarin was superior to rivaroxaban. 29 Similar results were borne by a meta-analysis of 12 studies comparing the NOACs (including edoxaban) to warfarin in nearly 55,000 patients, in which a 11% relative risk reduction was observed for both total and cardiovascular mortality. 30 Analysis of Danish public records encompassing over 13,000 propensity-matched patients showed that dabigatran was comparable to warfarin for prevention of stroke and systemic embolism and, over a median follow-up of 10.5 months, improved survival, prevention of pulmonary embolism, and was superior for intracranial hemorrhage, regardless of center TTR. 31 Most recently, a meta-analysis comprising the pivotal phase III studies for all four NOACs and totaling 71,683 patients, showed the NOACs to have significant reductions in stroke, intra-cranial hemorrhage and mortality with similar major bleeding (despite greater gastrointestinal bleeding), compared to warfarin (Figure 2). 32 The greatest relative reduction in major bleeding was noted amongst patients at centers with TTR <66%.
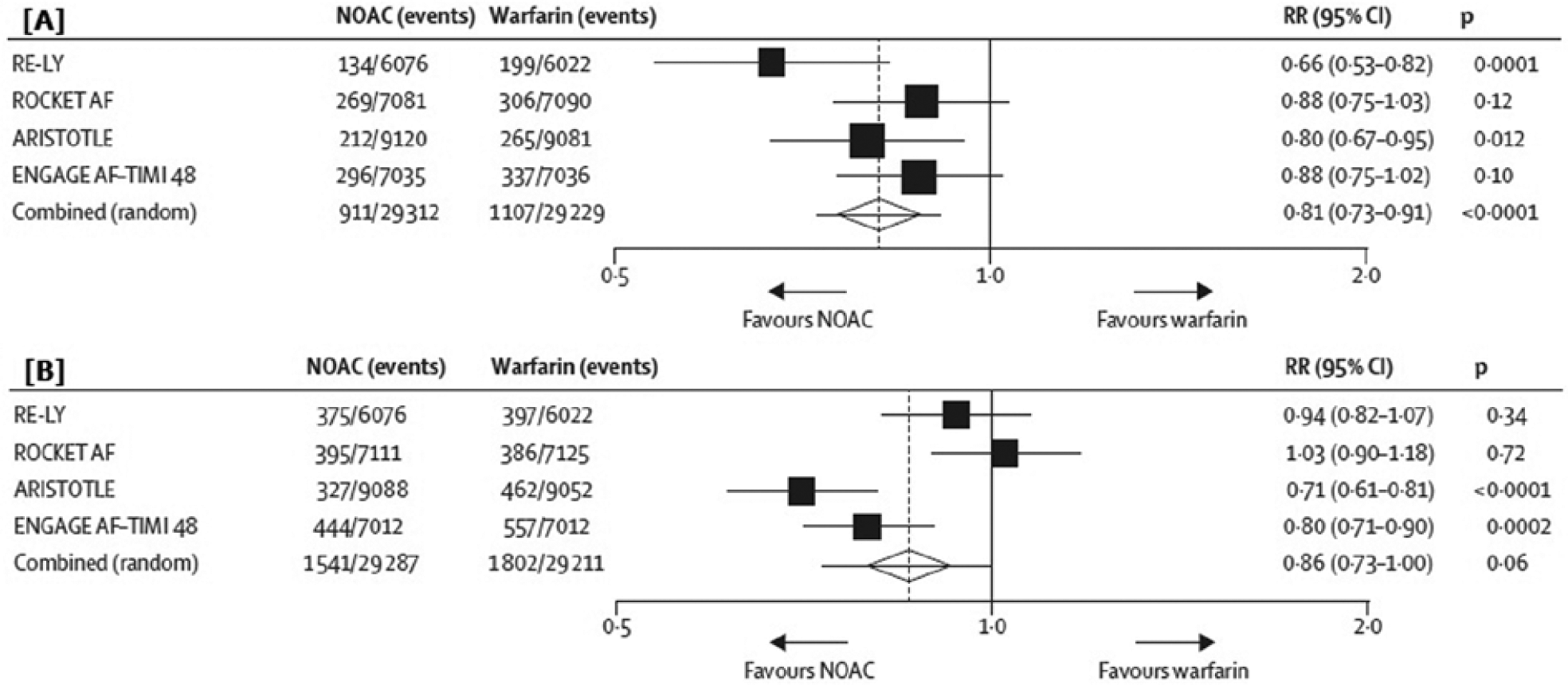
Efficacy and safety (major bleeding endpoints). (A) This Forrest plot demonstrates the efficacy of NOAC agents in major clinical trials compared to warfarin for stroke and systemic thromboembolic events. (B) Comparison of the NOAC agents for major bleeding events. Note: dosing is for dabigatran 150 mg twice daily, rivaroxaban 20 mg daily, apixaban 5 mg twice daily, and edoxaban 60 mg daily. Reprinted from The Lancet, 32 Copyright 2014, reproduced with permission from Elsevier.
The data thus far strongly suggest that for patients falling within tested bounds, the NOACs should be considered at least equally effective, safer than warfarin in terms of the intracranial hemorrhage and with the potential to improve mortality. Ultimately, the choice to switch should involve discussion between physician and patient that carefully addresses the relative risks and benefits. Shared decision-making ought to drive the decision to switch or not. In patients with additional risk factors for intracranial hemorrhage (ICH), it might be advantageous to switch to a NOAC, but this should not be recommended for all patients as absolute event rates are low and other considerations may apply.
Lack of antidotes
The lack of specific reversal agents for the NOACs has been a concern among prescribers and is a likely impediment to market uptake. Increasing reports of bleeding late in 2011 led to FDA and EMA (European Medicines Agency) reviews of both clinical trial data and post-marketing surveillance data, though no change in product labeling was ultimately recommended. Gradual reversal of warfarin has long been achieved by administering vitamin K, or in more emergent cases, fresh frozen plasma (FFP) or prothrombin complex concentrate (PCC). 33 NOAC reversal is indirectly achieved by withholding the drug, which has a relatively rapid rate of systemic clearance, and providing supportive care, which may include the administration of the aforementioned agents. PCC comes in three- and four-factor formulations, the latter including coagulation factors II, VII, IX, X, and proteins C and S. 34 Relative to FFP, PCC promotes faster normalization of the INR, more rapid and complete reversal of the warfarin effect, and can be infused rapidly in smaller volumes. 33 The two available three-factor formulations are not indicated for warfarin reversal and do not include factor VII, which is most rapidly depleted in warfarin-treated patients. 34 For patients with severe bleeding, the American College of Chest Physicians suggests four-factor PCC as the first choice for warfarin reversal (Grade 2C recommendation). However, prior to approval of the four-factor PCC product Kcentra in April 2013, formulations were not available in the US.35,36 Approval of Kcentra was based on a phase IIIb clinical trial of 216 warfarin-treated patients requiring urgent reversal because of major bleeding, randomized to receive vitamin K with FFP or vitamin K with PCC. 37 Cessation of major bleeding assessed 24 hours after the start of infusion was greater with Kcentra (72.4% vs 65.4%), as was INR reduction <1.3 (62.2% vs 9.6%). Additionally, infusion was completed seven times faster, and with 87% less volume. Because this was a relatively small trial, cost and apparent efficacy in clinical practice may be important determinants of market uptake, and FFP – which is approximately one-tenth as costly as PCC – may remain the mainstay for emergent warfarin reversal for some time to come. 38 Concern surrounding the lack of antidotes for the NOACs should be tempered by the ability to reverse the effect of warfarin in cases of severe bleeding, particularly ICH. Full reversal of INR with FFP may take up to 24 hours in patients on warfarin therapy.
Despite the availability of an ‘antidote’ for warfarin, the NOACs resulted in improved or equivalent rates of hemorrhage in major trials and were associated with an approximately 10% lower rate of all-cause mortality compared to warfarin. Subsequent analysis of the RE-LY trial revealed that spontaneous and traumatic intracranial hemorrhages are less common with dabigatran than with warfarin and morality rates after experiencing a stroke on-treatment are similar between groups. 39 A meta-analysis of five phase III trials including RE-LY also showed lower length of ICU stay and a trend toward lower mortality with all major bleeding on dabigatran compared to warfarin. 40 In addition, for patients undergoing invasive procedures, there was no difference in major bleeding or thromboembolic events between warfarin and dabigatran, and patients on dabigatran spent significantly less time off anticoagulation owing to its shorter half-life. 41 These findings suggest that the lack of a specific antidote does not compromise clinical outcome, particularly with ICH. Notably, PRT4445, an investigational drug described as a universal antidote for factor Xa inhibitors, has been developed. 42 A Fab fragment directed against dabigatran (aDabi-Fab) has shown excellent results in rat models and is currently under investigation in humans. 43 Although the management of bleeding events has never been compared prospectively, the security surrounding warfarin reversal and trepidation about the lack of reversal strategies for NOACs may be based more on perception than reality.
NOACs in patients with coronary artery disease
Warfarin reduces the risk of recurrent ischemic events in patients with coronary artery disease (CAD) when used alone or in combination with aspirin, but bleeding complications occur more frequently when anticoagulation is combined with a platelet inhibitor – particularly in patients on dual antiplatelet therapy. With the exception of edoxaban, each of the other three NOACs has been tested for safety in large phase II dosing-finding trials that included both STEMI and NSTEMI: dabigatran in RE-DEEM (Dose Finding Study for Dabigatran Etexilate in Patients With Acute Coronary Syndrome), rivaroxaban in ATLAS-1 ACS-TIMI 46 (Rivaroxaban in Combination With Aspirin Alone or With Aspirin and a Thienopyridine in Patients With Acute Coronary Syndromes–Thrombolysis In Myocardial Infarction 46), and apixaban in APPRAISE (Apixaban for Prevention of Acute Ischemic and Safety Events). The majority of patients in these studies were taking both clopidogrel and aspirin.
The RE-DEEM study of 1861 patients evaluated four dose regimens and showed a dose-dependent increase in clinically relevant bleeding with either 110 mg or 150 mg BID when given with dual antiplatelet therapy. 44 Gastrointestinal bleeding and epistaxis were the most common bleeding events. Although underpowered for efficacy, the composite of cardiovascular death, non-fatal MI, and non-hemorrhagic stroke were reduced in both higher-dose groups compared to lower doses of 75 mg and 50 mg BID or placebo.
ATLAS-1 ACS-TIMI 46 addressed two antiplatelet regimens: aspirin alone and dual-antiplatelet therapy in 3491 patients allocated to seven doses of rivaroxaban ranging from 5 mg to 20 mg daily. 45 A dose-dependent increase in clinically relevant bleeding on rivaroxaban was most pronounced in patients receiving concomitant dual antiplatelet therapy. Compared to placebo, rivaroxaban was associated with a significant decrease in death, MI, stroke and/or recurrent ischemia.
In APPRAISE-1, apixaban doses ranged from 2.5 mg BID to 20 mg once daily. 46 In 1715 patients, 76% on dual antiplatelet therapy, there was a dose-dependent increase in clinically relevant bleeding, and the highest doses (10 mg BID and 20 mg once daily) were terminated prematurely due to high bleeding rates. Subcutaneous bruising, epistaxis, hematuria, and GI bleeding were the most common hemorrhagic events reported. There was an insignificant trend toward reduction in cardiovascular death, MI, severe recurrent ischemia or ischemic stroke among those taking 2.5 mg BID or 10 mg once daily, with the greatest benefit in patients taking aspirin without clopidogrel.
Two phase III studies have been completed. APPRAISE-2 recruited 7392 patients with acute coronary syndrome (40% STEMI) and two additional risk factors for recurrent ischemic events, randomized to apixaban 5 mg BID (or 2.5 mg BID if CrCl was <40 ml/min) versus placebo. 47 The vast majority (81%) of patients were on dual antiplatelet therapy with aspirin and clopidogrel. The study was terminated after a mean follow-up of 241 days due to excess bleeding with apixaban compared to placebo (2.4 vs 0.9 events per 100 patient-years, HR=2.59, 95% CI 1.50–4.46) while there was no difference in the composite primary efficacy endpoint of cardiovascular death, myocardial infarction or ischemic stroke.
ATLAS-2 ACS-2-TIMI 51 (ATLAS-2) enrolled 15,526 patients randomized to placebo, rivaroxaban 2.5 mg BID or 5 mg BID. 48 Patients with prior GI bleeding, previous ischemic stroke, transient ischemic attack or impaired renal function were excluded from the highest dose arm. Over a mean of 13.1 months, both the 2.5 mg BID and 5 mg BID doses of rivaroxaban decreased the primary composite efficacy endpoint of cardiovascular death, MI or stroke compared to placebo (9.1% and 8.8%, respectively, for the two rivaroxaban doses vs 10.7% for placebo, p=0.02, 0.03). The 2.5 mg BID dose reduced all-cause and cardiovascular mortality, while the 5 mg BID dose reduced the rate of MI. Combined analysis of both doses revealed an increased risk of TIMI major bleeding vs placebo (2.1% vs 0.6%, p<0.001) and intracranial hemorrhage (0.6% vs 0.2%, p=0.009), without increasing fatal bleeding, though the 2.5 mg BID dose was associated with fewer fatal bleeds than the 5 mg dose (0.1% vs 0.4%, p=0.04).
The difference in efficacy outcomes across these phase III studies is intriguing, particularly as bleeding outcomes were similar. Early termination of APPRAISE-2 because of safety concerns handicaps interpretation, but the enrolled patient populations were dissimilar. Patients in APPRAISE-2 were older, had a higher prevalence of diabetes, chronic kidney disease, prior stroke, MI as opposed to unstable angina, and more NSTEMI rather than STEMI. Also, the 5 mg BID dose of apixaban in APPRAISE-2 was the same as for NVAF, while the doses of rivaroxaban used in ATLAS-2 were considerably lower than tested in the ROCKET AF study. Bleeding rates in patients receiving the factor Xa inhibitor were higher in ATLAS-2 than APPRIASE 2. The reduction in mortality in the group randomized to rivaroxaban 2.5 mg BID without reduction in MI, and the opposite effect in the 5 mg BID group, is difficult to understand. Finally, with regard to dabigatran, for which a phase III study has not been completed, the trends toward higher MI rates in RE-LY and reduction in MI in RE-DEEM leave unanswered whether the drug should be included in the discussion of therapy for patients at risk of coronary events.
Regardless of the study or drug, there was a three- to fourfold excess of major and intracranial bleeding in patients with acute coronary syndromes treated concurrently with NOACs and dual antiplatelet medication. The risk was more pronounced in populations at high risk of bleeding because of advanced age, female sex, low weight or impaired renal function. This was further evaluated in a meta-analysis of seven randomized placebo-controlled trials (including APPRAISE-2 and ATLAS-2) testing the NOACs in over 31,000 patients receiving antiplatelet therapy after an acute coronary syndrome. 49 Consistent with the major phase III studies, a threefold increase in major bleeding (OR 3.03, p<0.001) was noted. There is emerging evidence that NOACs may benefit specific subpopulations of ACS patients (unstable angina, STEMI or NSTEMI) or patients with other indications for anticoagulation who develop acute coronary syndrome. A recent analysis of the STEMI population in ATLAS-2 included 7817 patients stabilized after the index event (mean 4.7 days), nearly all of whom were on dual antiplatelet therapy. 50 Compared to placebo, rivaroxaban was associated with a reduction in the composite primary efficacy endpoint of cardiovascular death, MI or stroke (8.4% vs 10.6%, p=0.019), reaching significance within 30 days and remaining so over a 2-year follow-up period. This was largely driven by the 2.5 mg BID dosing group, which demonstrated marked reductions in cardiovascular death (HR=0.60, 95% CI 0.42–0.87) and all-cause mortality (HR=0.63, 95% CI 0.45–0.89). These end points were not affected in the 5 mg BID dose group. This was partly attributed to a better safety profile for the lower dose, which marginally increased intracranial hemorrhage (p=0.031) compared to the higher dose group (p=0.008). Comparing both doses to placebo, there was no difference in fatal bleeding (0.2% vs 0.1%, p=0.51). Based largely on the data from ATLAS-2, rivaroxaban 2.5 mg twice daily was approved by the European Commission for secondary prevention in adults with stabilized biomarker-confirmed acute coronary syndromes and is currently under FDA review. 51
Estimated from available evidence, the excess bleeding that occurs when dual antiplatelet therapy is combined with an anticoagulant is no lower with the NOACs than warfarin. The WOEST trial of 573 patients with CAD and indications for chronic anticoagulant therapy demonstrated that combined use of VKA and clopidogrel reduced bleeding compared to triple therapy with aspirin included (HR=0.36, 95% CI 0.26–0.50, p<0.001), while also reducing mortality. 52 The trial was open-label and the primary endpoint was the composite rate of minimal, minor and major bleeding according to the TIMI criteria, some of which is subjectively assessed. In line with the WOEST trial, registry data from Denmark demonstrated similar results. The registry examined patients with AF after MI or undergoing percutaneous coronary intervention (PCI) and found that single-agent clopidogrel with warfarin is comparable to triple therapy for prevention of VTE and outcomes of MI/coronary death, with a trend toward smaller bleeding risk. 53 Of note, OAC plus aspirin and aspirin plus clopidogrel were associated with a significant increased risk of all-cause death. The NOACs were not tested in this registry either. Considering the outcomes of APPRAISE-2 and ATLAS-2, in patients receiving aspirin alone compared to those on dual antiplatelet regimens, a potential role for therapy with a single antiplatelet agent, probably clopidogrel, along with one of the NOACs, is suggested. Selection of a dose that will maintain efficacy for prevention of thromboembolism in patients with advanced coronary disease who require anticoagulation for another reason without causing unacceptably high rates of bleeding is as yet an unmet challenge. In patients who require triple therapy, rivaroxaban may be the preferred agent as there was a trend toward lower rates of MI and statistically lower rates of intracranial hemorrhage. While studies to this point have been with clopidogrel, the P2Y12 inhibitors prasugrel and ticagrelor are increasingly used in place of clopidogrel as first-line agents and have not been tested with the NOACs. Both P2Y12 inhibitors were associated with reduced ischemic events (and lower mortality rates with ticagrelor) compared to clopidogrel, though associated with a minor increase in bleeding risk and concerns about safety in patients with previous stroke. In patients who require triple therapy, it may be beneficial to avoid prasugrel and ticagrelor, given the potential for increased risk of bleeding, though data assessing this strategy are lacking. It should also be noted that the risk reduction from adding rivaroxaban in the ATLAS-2 trial was similar to the risk reduction achieved with prasugrel in the TRITON-TIMI 38 trial and ticagrelor in the PLATO trial. Determining the optimal antithrombotic regimen for patients undergoing PCI requires factorial testing that addresses the increasing number of available antiplatelet options.
Off-label use and valve replacement
While marketing figures are hard to verify, by the last quarter of 2010 (a year after FDA approval), 92% of office prescriptions for dabigatran were for NVAF; a year later this figure fell to 63%. 54 Reasons for prescription varied, including VTE prevention, coronary disease, stroke or transient ischemic attack and, predominantly, hypertensive heart disease, which in the absence of AF is not an indication for any form of anticoagulation.
There were reports of mechanical heart valve thrombosis in patients switching between warfarin and dabigatran, 55 which prompted further investigation. The randomized, prospective RE-ALIGN trial compared warfarin with dabigatran for VTE in patients with mechanical heart valves. The study was halted early when it was found that patients receiving dabigatran were at increased risk of stroke, MI, valve thrombosis and bleeding compared to warfarin-treated patients, thus showing no benefit and increased risk. The authors postulated the failure of dabigatran could have been due to inadequate plasma levels postoperatively, alternative mechanisms of clot formation, and the broader spectrum of inhibition with warfarin – as it inhibits both the tissue factor pathway and the contact pathway-induced coagulation. 56 The FDA also noted that due to lack of appropriate testing, the use of dabigatran for bioprosthetic valves cannot be recommended. Trials evaluating the other agents for prosthetic valves are ongoing.
Given the established use of warfarin for patients with mechanical valves, consideration of NOAC use should be avoided until pertinent studies are completed. Short-term anticoagulation after bioprosthetic valve replacement has not been tested with the NOACs. Current recommendations are based on studies comparing warfarin and aspirin, which vary broadly in size, design, and outcomes. 57 This opens the door for future testing with NOACs to more definitively identify the safest and most effective therapy, particularly as advances in materials science bring forth newer, safer prosthetic valves and less thrombogenic suture material.
Lack of comparative effectiveness studies and choice of NOACs
Although large clinical trials have aptly compared NOACs with warfarin, no studies inform the decision of which NOAC to select for a given patient. Clinicians are left to draw upon data from pharmacological models and clinical trials with regard to dosing, adjustment for renal impairment, and side effects, and consider other clinical and socio-economic factors as well. However, differences in trial design, population characteristics, comparator uniformity, and definitions of efficacy and safety endpoints render comparison across studies inherently flawed. 58 For example, RE-LY was open-label and ROCKET AF recruited a higher risk population; furthermore, bleeding definitions, transitions of care, endpoint determination, and warfarin management differed. Without direct, unbiased, objective comparisons, decisions will remain open to the influence of subjective factors (including marketing). The impediments to developing such trials are overwhelming because financial incentives for companies that have invested billions of dollars over decades to develop, test and market a specific agent are inherently un-aligned with public interest. Public policy protects companies in the early phases of marketing newly approved drugs, and some have suggested a mandate for developers to set aside a portion of profit to support comparative studies, potentially in exchange for longer periods of initial proprietary protection. The challenge would then become assuring trial integrity and efficiency without bias. 59
Presently, extrapolation from primary trial data is the best available strategy for prescribing clinicians. In all phase III studies described above, intracranial hemorrhage – typically the most devastating complication of anticoagulation – occurred far less frequently with the NOACs than warfarin. Dabigatran causes dyspepsia, but was the only agent to demonstrate a reduction in ischemic stroke. Dabigatran, rivaroxaban and the higher dose of edoxaban were associated with more GI bleeding than warfarin, though apixaban was not. 60 Both rivaroxaban and apixaban are less dependent on renal elimination than dabigatran. Patients with a history of these specific features might lead the prescriber to choose the alternative option. Rivaroxaban and edoxaban have the benefit of once-daily dosing, which may be appealing for patients who are less compliant. Apixaban achieved superiority for both efficacy and safety endpoints, based mainly on lower risks of hemorrhage than warfarin.60,61 Certainly, though far fewer than with warfarin, drug interactions must be considered, given the interactions with P-gp inhibitors or inducers and the cytochrome P450 system. 1
Warfarin remains the drug of choice for patients with AF who have prosthetic heart valves or rheumatic heart disease. Patients with well-controlled INR values who have been stable may prefer to continue warfarin, viewing INR monitoring as an opportunity to connect with their provider. Warfarin will also remain the mainstay of therapy for patients with severe kidney disease, given its nearly complete hepatic metabolism.
Cost and market share
The US healthcare burden attributed to management of patients with AF is estimated between $3.2 and $13 billion annually and may rise to $30 billion by 2050 if the projected prevalence is sustained.62,63 This cost will be ultimately borne by the taxpayer, as both the incidence and risk associated with AF increase with age. Although warfarin is relatively inexpensive, ancillary monitoring, dose adjustment and complications can become costly. The price of the novel agents currently approved for marketing is approximately 10 times greater than warfarin, but a number of economic models and real-world studies of dabigatran suggest cost-effectiveness equal to or greater than warfarin for stroke prevention in patients with NVAF.60,64–67 Post-marketing cost-effectiveness analyses based on clinical use have not been completed for rivaroxaban or apixaban, and the market impact of edoxaban is yet to be seen. However, an event-specific cost analysis based on efficacy and safety endpoints encompassing the first three phase III AF studies estimated an annual per-person cost reduction of $179, $89 and $485 for dabigatran, rivaroxaban, and apixaban, respectively, relative to warfarin. 68 Cost reductions are driven primarily by reduction in hemorrhagic and ischemic stroke for dabigatran, reduction in hemorrhagic stroke and MI for rivaroxaban, and reduction in major bleeding including hemorrhagic stroke for apixaban. A Monte Carlo analysis testing the consistency of medical cost reduction estimates based on 10,000 random simulations drawn from Gaussian distributions around the means of each endpoint suggested cost reductions in 92.6%, 79.8% and 100% of simulations for dabigatran, rivaroxaban and apixaban, respectively. This analysis was based on trial data only and did not account for costs associated with poor treatment adherence, fluctuations in TTR, cost of warfarin monitoring and dose adjustment, or the fact that the warfarin efficacy in clinical practice is about half that achieved in clinical trials – suggesting even greater cost reductions with NOACs.
Theoretical modeling cannot entirely predict actual outcomes, and how much healthcare costs will be impacted once market uptake of the NOACs has plateaued remains uncertain. By the end of 2011, over a year after FDA approval, dabigatran had assumed an outpatient prescription rate of 19% for visits relating to oral anticoagulation, the remaining 81% involving warfarin. 54 For patients newly starting anticoagulation, the rate of dabigatran prescriptions appears to have stabilized at around 30–40%.69,70 When rivaroxaban entered the marketplace in November 2011, the proportion of prescriptions for warfarin remained unchanged, and those for rivaroxaban eroded the share previously occupied by dabigatran. It remains uncertain how the introduction of additional agents will alter this landscape.
Use in context of ablation or cardioversion
The thromboembolic risk associated with cardioversion for AF is 5–7%. This risk can be reduced by an order of magnitude with adequate anticoagulation. Whether by direct-current, pharmacological or ablative techniques, warfarin has long been the only oral anticoagulant approved for at least 3 weeks prior to and 4 weeks following cardioversion for AF. The NOACs have not been prospectively tested in this setting and there are no studies involving apixaban or edoxaban. However, post hoc analyses of patients in RE-LY and ROCKET AF are the only data published to inform the issue of use in the setting of cardioversion. In the RE-LY analysis, the number of patients in each of the three dose groups was similar. With or without trans-esophageal echocardiography, comparing both doses of dabigatran with warfarin, the study demonstrated no difference in stroke or systemic embolism (p=0.71 and 0.40 for 110 mg and 150 mg doses, respectively) or bleeding (p=0.06 and 0.99) at 30 days following cardioversion. This was despite lower rates of continuous treatment in both dabigatran dose groups over the 3 weeks prior to cardioversion, compared to warfarin (p<0.01 for both doses). These findings suggest that dabigatran can be reasonably used as an alternative to warfarin in this setting. Similarly, in a post hoc analysis of the ROCKET AF trial, there was no difference in the incidence of cardioversion between both groups (1.45/100 patient-years in the warfarin arm, and 1.46 in the rivaroxaban arm), and no significant difference in long-term rates of stroke, systemic embolism or survival in patients treated with rivaroxaban versus warfarin. 71 However, post hoc analyses should be interpreted with caution and further prospective studies should be performed to validate these findings.
In the setting of ablation, early studies have yielded varying results as to the safety of dabigatran therapy compared to continuous warfarin treatment. A recent prospective case–control study of 763 patients (191 in the dabigatran group) showed no difference in major or minor bleeding events, or pericardial tamponade between both groups when the drug was stopped at least 24 hours prior to the procedure. 72 Neither group experienced any thromboembolic events. In a similarly designed study of 290 patients, half of whom were taking dabigatran, there was a significantly greater major and total bleeding in the dabigatran group. 73 In this study, however, anticoagulation was discontinued on the day of the procedure. A third study showed that a strategy of withholding only the dose prior to ablation, and restarting immediately after sheaths are removed or the patient is transferred to the medical monitoring unit, did not yield differences in hemorrhagic (1.2% vs 1.5%, p=0.74) or thromboembolic (3.2% vs 4.1%, p=0.53) complications amongst propensity-matched cohorts of 344 patients each. 74 A systematic review and meta-analysis by Providência et al. involving 4782 patients undergoing catheter ablation again showed no difference between dabigatran and warfarin in clinical endpoints of systemic thromboembolism or major bleeding, though overall event rates were low. 75 A recent meta-analysis of 1113 patients comparing rivaroxaban to warfarin in this same context also showed no difference in systemic embolism or bleeding. 76
Based on these results, dabigatran, and perhaps rivaroxaban and other NOACs, can be safely and efficaciously administered in the context of ablation, though adequately powered, randomized, prospective studies to validate this strategy are lacking.
Management of events during NOAC therapy
On-treatment events present challenging management scenarios, as there are no large-scale randomized trials comparing treatment strategies for either ischemic or hemorrhagic complications. Data from RE-LY showed a non-significant increase in non-fatal MI compared with warfarin (annual rates of 0.82% and 0.81% with dabigatran 110 mg or 150 mg BID compared with 0.64% with warfarin. 77 In the warfarin group, serious bleeding (regardless of INR) was treated with 10 mg of intravenous vitamin K, and FFP, prothrombin concentrate complex or recombinant factor VIIa depending on clinical urgency. 78 In ARISTOTLE, the study protocol provided guidelines for management of bleeding events including discontinuation of antithrombotic therapy, local hemostatic measures and administration of FFP as needed. 79 Moreover, they noted that other agents, including recombinant activated factor VII, had not been well studied and were not recommended. Neither RE-LY nor ARISTOTLE offered a pre-specified management strategy for thrombotic events. Pre-specified strategies for management of thrombotic and hemorrhagic events were not provided in ENGAGE AF.
In ROCKET AF, guidance was provided regarding management of both thrombotic and hemorrhagic events. Patients undergoing vascular interventions, including PCI, had their study drugs continued and could receive dual antiplatelet therapy with aspirin and a thienopyridine at the investigator’s discretion. 80 For patients with bleeding complications, management strategies followed local standards of care, though these were not discussed in detail and likely varied from site to site. With all procedures, the study drug was resumed when hemostasis was achieved and the treating physician considered oral anticoagulant therapy appropriate.
Broadly, management of thrombotic events on the NOACs should be similar to that for patients taking warfarin: drawing attention to adherence and assessment of patient-specific bleeding risk to minimize complications of antithrombotic therapy. It is reasonable to switch from a NOAC to warfarin given the ability to monitor levels, familiarity and ease of use, and broader spectrum of anticoagulation, as warfarin affects both the intrinsic and extrinsic pathways. Aspirin may be added in accordance with the current guidelines and based on individual patient characteristics.
Patients experiencing hemorrhagic complications on NOACs should be treated with supportive care, particularly given the short half-life of these agents and the present lack of specific antidotes. Early consultation with hematology experts should be strongly considered. With minor bleeding complications, doses of these agents may be held until hemostatic control is achieved. With moderate to severe bleeding, administration of FFP, vitamin K or PCC may be warranted, though, as noted earlier, no data regarding clinical efficacy are available. A meta-analysis of five phase III clinical trials of dabigatran showed that patients who experienced major bleeding events had a trend toward lower mortality with dabigatran over warfarin, though this did not reach statistical significance. They also had shorter ICU stays and required more red blood cell transfusion but less plasma (Figure 3). 40
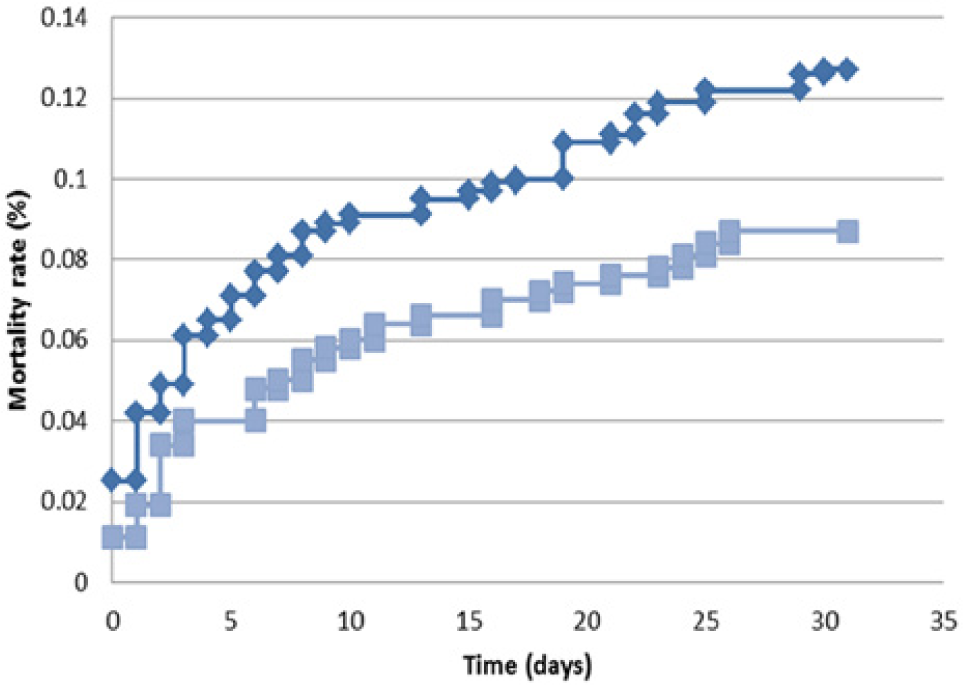
Thirty-day mortality after a major bleeding event. Adapted with permission from Circulation. 40 This data was obtained from the RE-LY database. Warfarin is depicted in dark blue. Dabigatran is depicted in light blue.
Procoagulant agents, such as PCC, activated prothrombin concentrate complex (aPCC), recombinant factor VIII (FEIBA) and recombinant factor VIIa had previously been evaluated in small-scale trials involving healthy volunteers, animal models and in vitro studies, lacking clinical outcomes data. 81 Recently, there was a multinational, randomized, open-label trial comparing four-factor PCC and plasma for treatment of major bleeding events in patients on VKA therapy. Though the trial was small, was not powered for major clinical endpoints, and did not examine the type of VKA therapy, four-factor PCC was found to be non-inferior to plasma for hemostatic efficacy and actually superior for rapid reversal of INR in the setting of major bleeding events. 82
The 2013 European Heart Rhythm Association guidelines recommend treating life-threatening bleeding according to accepted clinical practice by controlling hemostasis, administering FFP (as a volume expander) and transfusing platelets as needed. Consideration of PCC at a dose of 25 units/kg or activated PCC 50 IE/kg is also recommended, despite the lack of clinical data supporting this or evidence of efficacy of one treatment over another. 83 When there is an unacceptably high risk of thrombosis or clinical urgency and the patient has a hemorrhagic complication on one of the NOACs, switching to warfarin with close monitoring of the INR – particularly in patients with new or worsening renal failure – may be advisable. However, there are scant data to support this recommendation and the INR may not accurately reflect the true level of anticoagulant activity. A post hoc analysis of the ROCKET AF trial suggests that rivaroxaban-treated patients discontinuing or interrupting therapy are at increased risk of stroke and systemic embolism compared with warfarin-treated patients, suggesting a role for therapeutic anticoagulation during periods of treatment interruption. 84
The question of rebound thrombogenicity
The concept of rebound thrombosis following withdrawal of anticoagulation has been debated for nearly as long as warfarin has been on the market. Proponents who previously described an increase in thrombotic events after stopping warfarin are questioning whether a more robust manifestation of this phenomenon could occur following NOAC interruption or while switching to the NOACs from other agents.
Analysis of prescribing patterns in Denmark during the 4 months following dabigatran approval showed higher rates of thromboembolic and hemorrhagic complications in patients switched from warfarin to dabigatran compared to those continuing warfarin, 85 a pattern not observed in patients newly started on dabigatran. This may be due in part to inappropriate dosing, unfamiliarity with dosing during the crossover period, off-label use or attempts to switch patients who were inherently challenging to control or were followed less closely than necessary.
Notably, in the major clinical studies, a large proportion of patients were warfarin-naïve: 50% in RE-LY, 37.6% in ROCKET AF, 43% in ARISTOTLE, and 41% in ENGAGE AF.14,58 The results of both ROCKET AF, ARISTOTLE and ENGAGE AF can be thought to reflect a rebound effect, as overall end point determination included the transition from the study drug back to warfarin. In RE-LY, however, the switch back to warfarin was not standard to the study protocol, and as such the impact of drug cessation on clinical events cannot be elucidated.
In a post hoc analysis of ROCKET AF, the incidence of stroke or non-CNS embolism within 30 days of (i) temporary interruptions of 3 days or more, (ii) early permanent study drug discontinuation, or (iii) end-of-study transition to open-label therapy was compared to patients who did not have any discontinuations. Among 7061 rivaroxaban-treated patients in ROCKET AF, there were 4021 such discontinuations vs 4240 of 7082 warfarin-treated patients. Event rates were similar after temporary interruptions and early permanent discontinuation. Patients transitioning from rivaroxaban to warfarin at the end of study, however, suffered more strokes (6.42 vs 1.73/100 patient-years, HR=3.72, 95% CI 1.51–9.16, p=0.0044) and more thromboembolic (stroke, non-CNS embolism, MI and vascular death) events (9.05 vs 4.03/100 patient-years, HR=2.24, 95% CI 1.19–4.22, p=0.012).
Data such as these suggest that additional coverage should be provided to patients transitioning from rivaroxaban to warfarin, particularly the relatively high-risk patients enrolled in ROCKET AF, but such coverage during transitions has not been tested. Additionally, logistical and pharmacokinetic factors affecting therapeutic anticoagulation during transition may contribute to rebound phenomena. Analysis of the 30-day transition period after ROCKET AF showed that while 83% of the warfarin group had achieved one or more therapeutic INR, only 52% of rivaroxaban-treated patients achieved the same (p<0.0001), 86 and the median time to reach therapeutic INR was 3 days in the warfarin-treated group vs 13 days in the rivaroxaban group. These data suggest that an inherent rebound thrombogenic state following cessation of anticoagulation is less likely than exposure of an inherent risk of thromboembolism. Furthermore, the magnitude of rebound after interruption of the NOACs compared to warfarin has not been comparatively investigated.
Renal function and elderly patients
Elderly patients are more likely to experience fluctuations in renal function, have poly-pharmacy, have a history of bleeding, undergo hospitalization, suffer falls, and display cognitive impairment that could lead to over- or under-dosing. Along with the gradual decrease in glomerular filtration rate (GFR) and increase in risk of tubular-interstitial injury known to be associated with aging, these factors have raised concerns about NOAC use in the elderly. Although reduced doses of dabigatran, rivaroxaban and edoxaban are available for elderly patients with CrCl 30–59 ml/min, 15–29 ml/min and 30–50 ml/min, respectively, patients with more advanced renal dysfunction were excluded from RE-LY, ROCKET AF and ENGAGE AF and relatively few patients in the AVERROES and ARISTOTLE trials actually received the reduced dose of apixaban. Pharmacokinetic studies of dabigatran demonstrated marked differences in plasma concentrations over time spanning the full spectrum of renal impairment. A subgroup analysis of RE-LY showed that compared to patients with normal renal function, those with mild and moderate renal dysfunction had proportionately higher rates of stroke and systemic embolism. Nevertheless, both the efficacy and safety of dabigatran were independent of age and renal function. A subgroup analysis of ROCKET AF showed that patients with impaired renal function were generally sicker than the general study population, and had higher event rates irrespective of treatment, but experienced the same relative effects of rivaroxaban compared to warfarin as in the overall trial population. These findings support the stance that bleeding risk is higher in patients with renal impairment regardless of agent and that the lower intracranial bleeding risk conferred by NOACs in clinical trials should be regarded favorably for elderly patients, particularly in the setting of equivalent or superior efficacy. The superiority of apixaban was maintained across the spectrum of renal function tested, and patients with a GFR ≤50 ml/min had the greatest reduction in bleeding compared to warfarin (HR=0.50, 95% CI 0.38–0.66; interaction p=0.005). 87 At this time, there are no clinical data on how to approach patients with fluctuating renal function on NOACs, though it has been suggested that these agents be stopped and high-risk patients bridged with heparin, but this method has not been tested.
Conclusion
Although the NOACs met considerable anticipation and excitement, multiple questions remain about their safety and efficacy in broad clinical use outside the context of clinical trials. Additionally, data are lacking to inform selection of one agent over another. Although enthusiasm for these agents remains strong, warfarin is still the most frequently prescribed oral anticoagulant. The rate at which warfarin is replaced by new oral anticoagulants will depend on clinical experience and how well patients tolerate these drugs, the quality of new data that emerge from further analysis of available databases, reimbursement policies and other market forces.
Footnotes
Declaration of conflicting interest
Dr Halperin receives honoraria from Johnson & Johnson and Bayer, and advisory fees from Boehringer Ingelheim, Bristol-Myers Squibb, and Pfizer. None of the other authors have any disclosures or conflicts of interests to declare.
We confirm this manuscript has not been published in any other location and is not under review by any other medical journal. All authors participated in the creation of this manuscript, approved its final version and agree with its submission.
Funding
This research received no specific grant from any funding agency in the public, commercial, or not-for-profit sectors.