
Abstract
Nitrite stores decrease after exercise in patients with peripheral artery disease (PAD) and diabetes represents decreased nitric oxide (NO) bioavailability that may contribute to endothelial dysfunction and limit exercise duration. The primary objective of this placebo-controlled study was the safety and tolerability of multiple doses of oral sodium nitrite in patients with PAD, predominantly with diabetes, over a period of 10 weeks. The primary efficacy endpoint was endothelial flow-mediated dilatation (FMD) and secondary efficacy endpoints included a 6-minute walk test and quality of life assessment. Of the 55 subjects, the most common side effects attributed to sodium nitrite were a composite of headache and dizziness occurring in 21% with the 40 mg dose and 44% with the 80 mg dose. There was no clinically significant elevation of methemoglobin. FMD non-significantly worsened in the placebo and 40 mg groups, but was stable in the 80 mg group. Diabetic patients receiving 80 mg had significantly higher FMD compared with the placebo and 40 mg groups. There was no significant change in 6-minute walk test or quality of life parameters over time compared to placebo. In conclusion, sodium nitrite therapy is well tolerated in patients with PAD. The possible clinical benefit of sodium nitrite should be studied in a larger and fully powered trial.
Introduction
Peripheral artery disease (PAD) is a manifestation of systemic atherosclerosis and is a strong predictor of cardiovascular (CV) mortality.1–3 During exercise, peripheral limb arterial stenoses limit the ability to appropriately increase blood flow, which leads to an oxygen supply/metabolic demand mismatch, a bio-energetic deficit, and subsequent muscle contractile dysfunction with the associated symptoms of claudication. Thus, the primary pathophysiology of PAD is related to the limitation in blood flow and abnormal hemodynamics (reduced tissue perfusion pressure and blood flow) during exercise.
A consistent feature in vascular diseases, as well as diabetes, is dysfunction in nitric oxide (NO)-dependent signaling processes, occurring either through a deficit in NO synthesis, NO bioavailability, or both. 4 Clinically, endothelial NO bioactivity can be assessed non-invasively using an ultrasound-based technique of flow-mediated dilation (FMD) of the conduit arteries, and endothelium-derived NO production is reduced in diabetes and PAD. 5 Importantly, abnormal endothelial function and reduced arterial NO bioavailability is independently associated with adverse long-term outcomes in several populations, and also in patients with PAD. 6
Recently, interest has focused on non-enzymatic sources of NO that may be amenable to therapeutic manipulation. For example, the metabolic products of NO metabolism such as nitrite and nitrate, once thought of as NO metabolism end products, may serve as an alternative source of NO under certain physiological conditions, such as hypoxia and ischemia.7–9 Nitrite is a first-order metabolite of NO oxidation and a marker of constitutive NOS activity. More recently, nitrite has been advanced as a circulating NO storage depot and delivery source, reacting with oxyhemoglobin to form nitrate and methemoglobin (MetHb) or with deoxyhemoglobin to form NO, nitrosyl hemoglobin, and other NO adducts. 10 Because nitrite is found ubiquitously in the systemic circulation, the dual fates of nitrite metabolism position it as a unique physiological source of NO in pathophysiological conditions such as tissue ischemia. Circulating plasma nitrite levels are reduced in patients with diabetes and PAD. 11
Nitrite stores decrease following exercise among individuals with PAD, with and without diabetes (40% reduction), compared with healthy individuals.12,13 These results are consistent with use of NO stores in an attempt to normalize blood flow and oxygen delivery. As nitrite stores become depleted, the resulting loss of NO may contribute to abnormal endothelial function and circulatory responses in PAD. Moreover, it has been recently reported that nitrite therapy prevents Db/Db diabetic mouse hind limb ischemic tissue injury in an NO-dependent manner. 14
Restoring NO bioavailability may therefore offer a therapeutic opportunity. Although many structurally and chemically diverse NO-donor compounds have been synthesized, such as nitroglycerin and nitroprusside, they are limited in their ability to deliver NO to specific sites. A potential consequence is that NO-donors elicit systemic vascular effects resulting in hypotension. Because of the selective nature of nitrite metabolism and its ubiquitous nature, supplemental nitrite is uniquely positioned as a NO-donor for treatment of cardiovascular diseases.
The primary goal of this dose-ranging study was to evaluate the safety, pharmacokinetic profile and tolerability of multiple doses of oral sodium nitrite in patients with diabetes and PAD. Efficacy measures included vascular reactivity, walking ability and lifestyle assessment. The hypothesis tested was that oral sodium nitrite is safe and improves the efficacy measures listed above.
Methods
Study design and participants
This was a randomized, double-blinded, placebo-controlled, phase IIa dose-ranging study involving 10 study sites. The primary objective was to assess the safety and tolerability of multiple doses of twice-daily 40 mg and 80 mg sodium nitrite compared with placebo over a 10-week treatment period. Secondary objectives were to evaluate endothelial function, and markers of functional improvement and biologic activity, including: the 6-minute walk distance, quality of life (Walking Impairment Questionnaire (WIQ) and RAND 36) and selected biomarkers.
Inclusion criteria
Subjects with PAD and an ankle–brachial index < 0.90 who were symptomatically stable for at least 1 month were enrolled. Subjects were between the ages of 35 and 85 years old and women had to be post-menopausal, sterilized or using suitable birth control. The initial protocol required that patients also have diabetes mellitus Type I or Type II requiring either oral therapy or insulin that had been present for at least 3 months. If not medically treated, subjects had a HbA1c ≥ 6.5% on two separate occasions of fasting blood glucose levels of at least 126 mg/dL after an 8-hour fast. However, owing to recruitment challenges, the protocol was amended to include subjects with PAD who did not have diabetes.
Exclusion criteria
The exclusion criteria included patients with non-atherosclerotic PAD (e.g. Buerger’s or vasculitis); lower extremity surgical or percutaneous revascularization within the last 6 months or anticipated within the treatment period; myocardial infarction, unstable angina, cerebrovascular accident or transient ischemic attack (TIA) within the last 3 months; poorly controlled diabetes (HbA1c > 10.0%); and poorly controlled hypertension (systolic blood pressure (SBP) ≥ 160 mmHg or diastolic blood pressure (DBP) ≥ 100 mmHg) despite therapy. In addition, patients were excluded for SBP ≤ 100 mmHg on current medical regimen, hypersensitivity to sodium nitrite or related compounds, renal insufficiency documented as estimated glomerular filtration rate (eGFR) < 90 mL/minute/1.73 m2 (Modification of Diet in Renal Disease Study (MDRD)), and any pregnant or nursing women. Other exclusion criteria included active malignancy, active infection (i.e. systemic or osteomyelitis), class III or IV heart failure, a diagnosis of critical leg ischemia (CLI), previous amputation or planned amputation within 3 months, the subject’s ability to perform the 6-minute walk test limited by symptoms other than claudication, history of MetHb (> 1.5 g/dL), evidence of anemia, history of a chronic hemolytic condition – including sickle cell disease, use of anti-migraine medication such as Imitrex (sumatriptan), glucose-6-phosphate dehydrogenase (G6PD) deficiency on screening, abnormal hemoglobin electrophoresis at screening and regular use of: allopurinol, tricyclic antidepressants, antihistamines, meperidine and related central nervous system (CNS) depressants, and nitrates.
Treatments administered
After signing informed consent approved by the respective study site investigational review board, the subjects underwent a screening visit. If the subjects met the study criteria, they were randomized (1:1:1) to placebo, 40 mg BID or 80 mg BID sodium nitrite (Figure 1). All doses were given as twice-daily oral pills for 10 weeks. Following the 10-week dosing period, patients in each treatment arm entered a 1-week dose-escalation period (dose-doubling). Patients in the 40 mg sodium nitrite BID group dose-escalated to 80 mg sodium nitrite BID for 7 days, and patients in the 80 mg sodium nitrite BID group dose-escalated to 160 mg sodium nitrite BID for 7 days. Subjects taking placebo doubled the number of placebo capsules taken BID. All study medications were stopped at the end of the 7-day dose-escalation period.
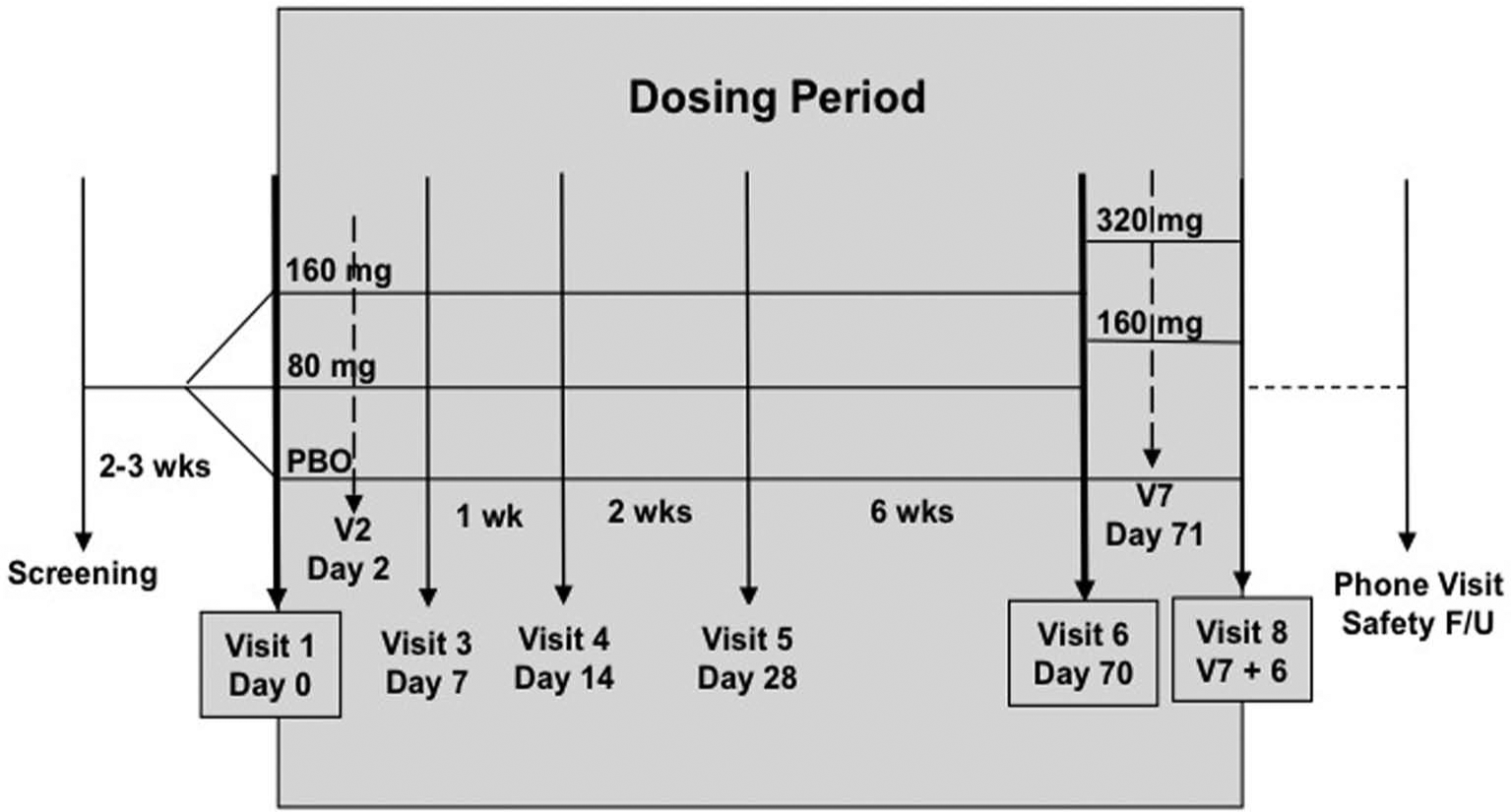
The study design depicting the dosing period over the study course. (PBO, placebo; wks, weeks; F/U, follow-up.)
Efficacy parameters
Flow-mediated vasodilation of the brachial artery was assessed according to previously published protocols. 5 In brief, a blood pressure cuff was applied to the upper arm and inflated to suprasystolic pressure for 5 minutes. The diameter of the brachial artery was assessed at baseline before cuff inflation and at 1 minute after cuff deflation. A 6-minute walk was performed using a standardized protocol. This was performed on a 50-foot (15.24-meter) straight course with no obstacles with visible distance markers. The patient was instructed to walk as far as possible in 6 minutes (even if claudication developed) and the distance walked at the end of 6 minutes was recorded (in feet). Quality of life was measured by two questionnaires – WIQ and RAND 36 – which were completed prior to the 6-minute walk test.
Safety parameters
The following safety parameters were assessed: medical and medication history, concomitant medication usage, physical examination, vital signs, 12-lead electrocardiogram (ECG), clinical chemistries, complete blood count (CBC), coagulation studies, urinalysis, and adverse events. MetHb was measured in the local laboratory of the study site at seven time points starting at the first visit. Observation for development of severe adverse events (e.g. decrease in blood pressure, dizziness) was done during the first 4 hours after administration of the first dose at each dose level of sodium nitrite. Subjects underwent measurement of blood pressure and heart rate lying and after standing for 5 minutes.
Glutathione and glutathione disulfide
The measurement of glutathione (GSH) and glutathione disulfide (GSSG) levels was performed as previously reported. 15 After 100 mM N-ethylmaleimide (NEM) treatment, the mixtures were diluted with an equal volume of trichloroacetic acid (TCA) (10%) and then centrifuged at 12,000 rpm for 5 minutes. Excess NEM in the supernatant was extracted by 5 volumes of dichloromethane. After centrifugation, the supernatant was alkalinized with a few drops of 1 M Tris–HCl (pH 10), and then reacted with an equal volume of a 1-fluoro-2,4-dinitrobenzene (DNFB) solution (1.5% in ethanol) for 3 hours at room temperature in the dark. After acidification (pH < 5 with 37% HCl), samples were analyzed by reversed-phase high-performance liquid chromatography (RP-HPLC) with a NH2 column and a UV detector at 355 nm. Concentrations of GSH and GSSG were calculated using standard calibration curves.
Arginine, homoarginine, symmetric dimethylarginine and asymmetric dimethylarginine
The measurement of arginine, homoarginine, symmetric dimethylarginine (SDMA) and asymmetric dimethylarginine (ADMA) was performed as previously published. 16 Samples were spiked with monomethylarginine as an internal standard and purified by solid-phase extraction on Oasis® MCX SPE columns. Thereafter, the samples were dried by evaporation with nitrogen at a temperature of 60–80°C and derivatized with ortho-phthaldialdehyde reagent containing 3-mercaptopropionic acid. Detection was performed with fluorescence detection (ex: 340 nm and em: 455 nm) by RP-HPLC.
Total cysteine, total homocysteine and total GSH
The measurement of total cysteine, total homocysteine and total GSH was performed as per the previously published method. 17 Samples were reduced by the addition of Tris(2-carboxyethyl)phosphine hydrochloride (TCEP) and left at room temperature for 30 minutes. To precipitate proteins, 100 g/L TCA was added. After centrifugation for 10 minutes, the supernants were derivatized with 4-fluoro-7-sulfobenzofurazan (SBD-F). The fluorescent thiol derivatives were separated on a C18 column using isocratic elution by RP-HPLC with a fluorescence detector (ex: 385 nm, em: 515 nm).
Total sulfide
The measurement of total sulfide was performed as per the previously published method.18,19 Samples were incubated with TCEP in 100 mM phosphate buffer (pH 2.6, 0.1 mM DTPA) in volatilized H2S and subsequently trapped with 100 mM Tris–HCl (pH 9.5, 0.1 mM DTPA) and then reacted with excess monobromobimane. The fluorescent product sulfide-dibimane (SDB) was separated on an C18 column using isocratic elution by RP-HPLC with a fluorescence detector (ex: 390 nm, em: 475 nm).
Statistical analysis
Descriptive statistics were used to summarize the demographic and clinical data, such as ECGs and vital signs. Laboratory values above and below the normal limit were identified, and adverse events presented by System-Organ Class, severity and relationship to study treatment.
The primary efficacy analysis was comparison of the change in FMD from baseline between the pooled drug and placebo treated groups following 10 weeks of treatment using an unpaired t-test. In case of a substantially skewed distribution within the comparison groups, a non-parametric two-sample Wilcoxon signed-rank test was used. For dichotomized efficacy endpoints the null hypothesis H0: rc=rp versus H1: rc>rp was to be tested, where rc is the proportion of subjects with improved results in the treated cohort and rp is the proportion of subjects with improved results in the placebo cohort. The difference between proportions in each of the groups (drug versus placebo and low dose versus high dose) was tested with chi-squared test or Fisher’s exact test. Secondary analyses employed repeated-measures analysis of variance (ANOVA) based on generalized estimating equations to incorporate time, group and interaction. Analysis of the secondary endpoints such as the 6-minute walk test and quality of life questionnaires were performed using the least-squares means, standard errors, and p-values based upon an analysis of covariance (ANCOVA), with a fixed effect of treatment group, and Day 0 (baseline) as a covariate. All statistical decisions were made before un-blinding. Functional parameters were tabulated by dose and overall. Summary statistics were computed and log dose–response curves prepared for each parameter as appropriate. The primary analysis population was intention-to-treat defined as all patients receiving a dose of study drug and having a follow-up efficacy visit. A secondary prospectively defined population was the large subgroup of subjects with diabetes.
Sample size calculation
With a total sample size of 34 subjects in the pooled sodium nitrite group and 16 subjects in the placebo group, the study had 82% power to detect a difference of 1.4% in FMD between the pooled sodium nitrite treated patients compared with placebo at the 0.050 two-sided level of significance.
Results
Clinical characteristics
Subjects were predominantly male and white, but there were no significant differences in the risk factor profile or severity of PAD (as measured by the ABI) between the three groups (Table 1). Drug exposure defined as days on study drug and study drug adherence was similar between the three groups (Table 2).
Baseline characteristics.
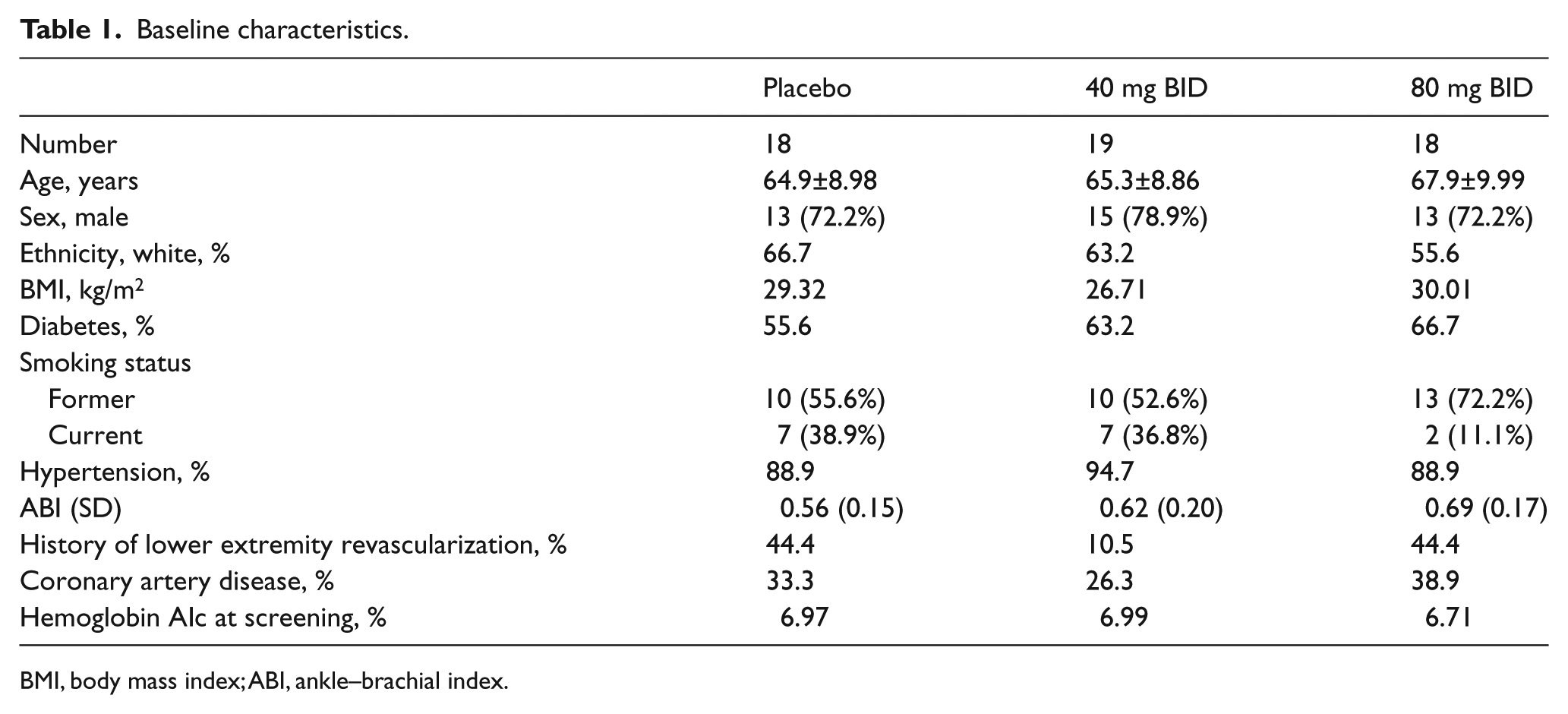
BMI, body mass index; ABI, ankle–brachial index.
Drug exposure and summary of subject disposition.
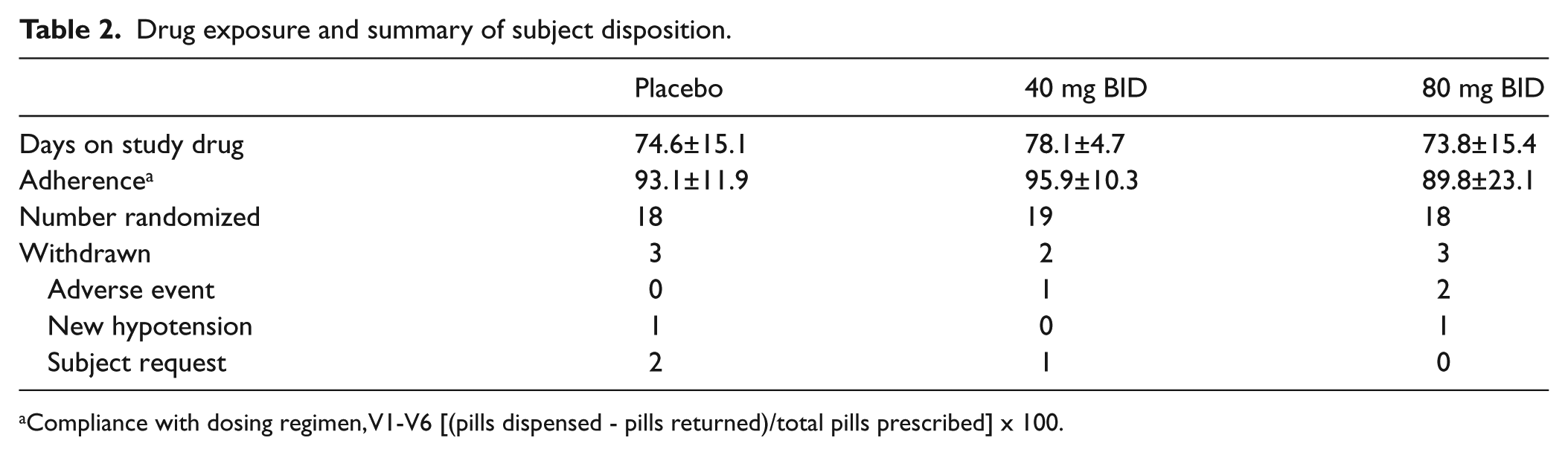
Compliance with dosing regimen, V1-V6 [(pills dispensed - pills returned)/total pills prescribed] x 100.
Safety data
Sodium nitrite was well tolerated, with a similar withdrawal rate between groups (Table 2). The number of subjects with an adverse event was numerically higher in the 40 and 80 mg groups compared with placebo (Table 3). There were two serious adverse events on placebo but none in either of the drug dose groups (Table 3). Headache and dizziness were the most common adverse events attributed to study medication, occurring in four patients in the 40 mg BID group and eight patients in the 80 mg BID group (Table 3). There were no imbalances in gastrointestinal disorders (numerically more on placebo), or in investigations, or musculoskeletal, respiratory, vascular or skin disorders. There was one renal failure noted on the 40 mg dose.
Safety data and summary of adverse events.
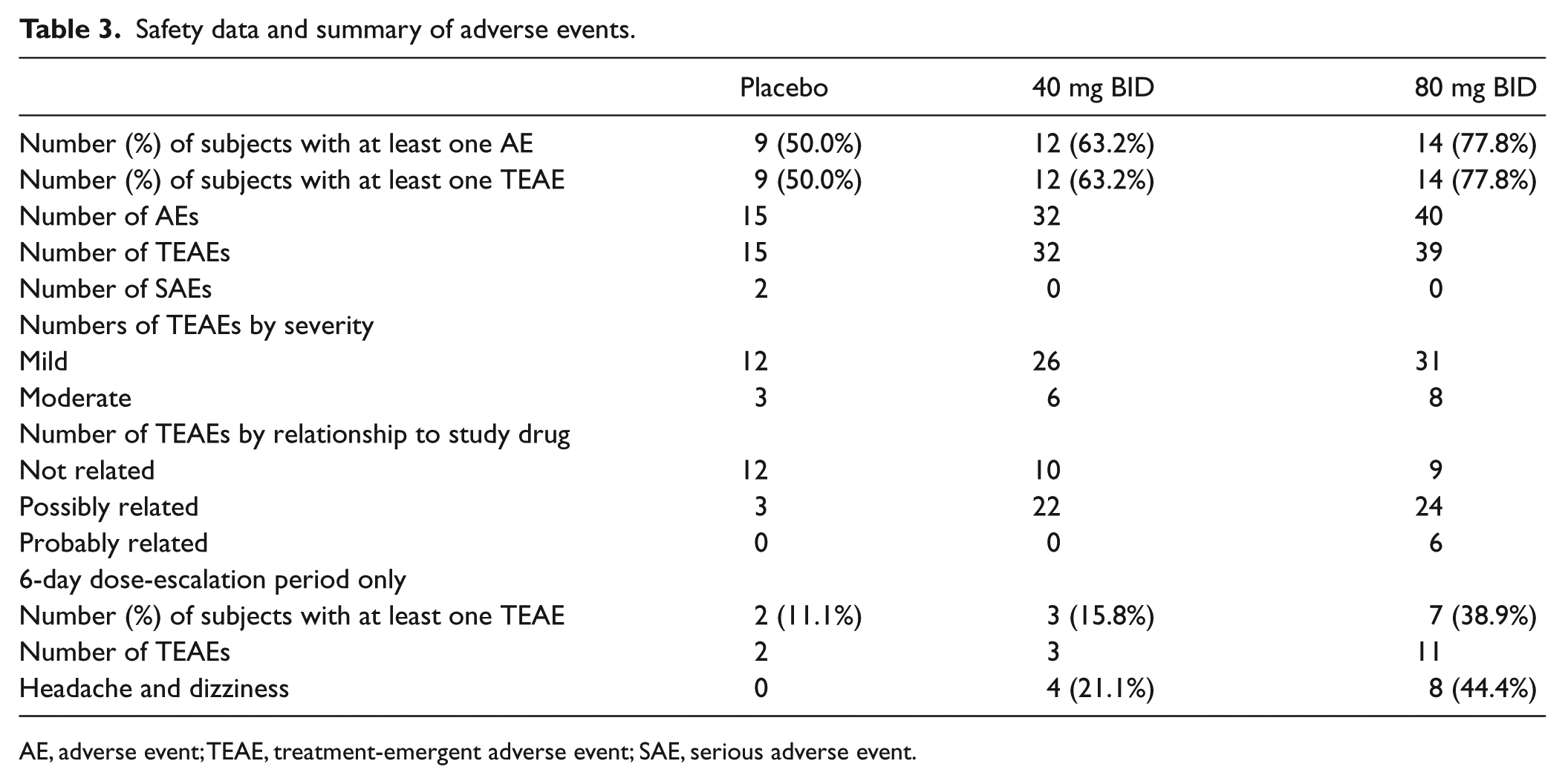
AE, adverse event; TEAE, treatment-emergent adverse event; SAE, serious adverse event.
Safety laboratory data were evaluated as shift plots. The data revealed no imbalances between groups in shifts from low or normal to high values for liver function tests or in creatinine levels.
Nitrite reacts with oxyhemoglobin to form nitrate and MetHb or with deoxyhemoglobin to form NO, nitrosyl hemoglobin and other NO adducts. MetHb remained stable throughout the study period in subjects randomized to placebo and those randomized to the 40 mg BID dose (Figure 2). However, when the 40 mg BID group increased their dose to 80 mg at week 6 the MetHb values increased approximately 0.55% at 30 minutes post dose. In the 80 mg BID group, MetHb increased 0.82% at 1 hour post dose then declined at subsequent time points. When the 80 mg BID group increased their dose to 160 mg BID, at 30 minutes post dose the MetHb increased by 2.11%. Overall, changes in MetHb due to any dose regimen of nitrite never surpassed 3% total MetHb.
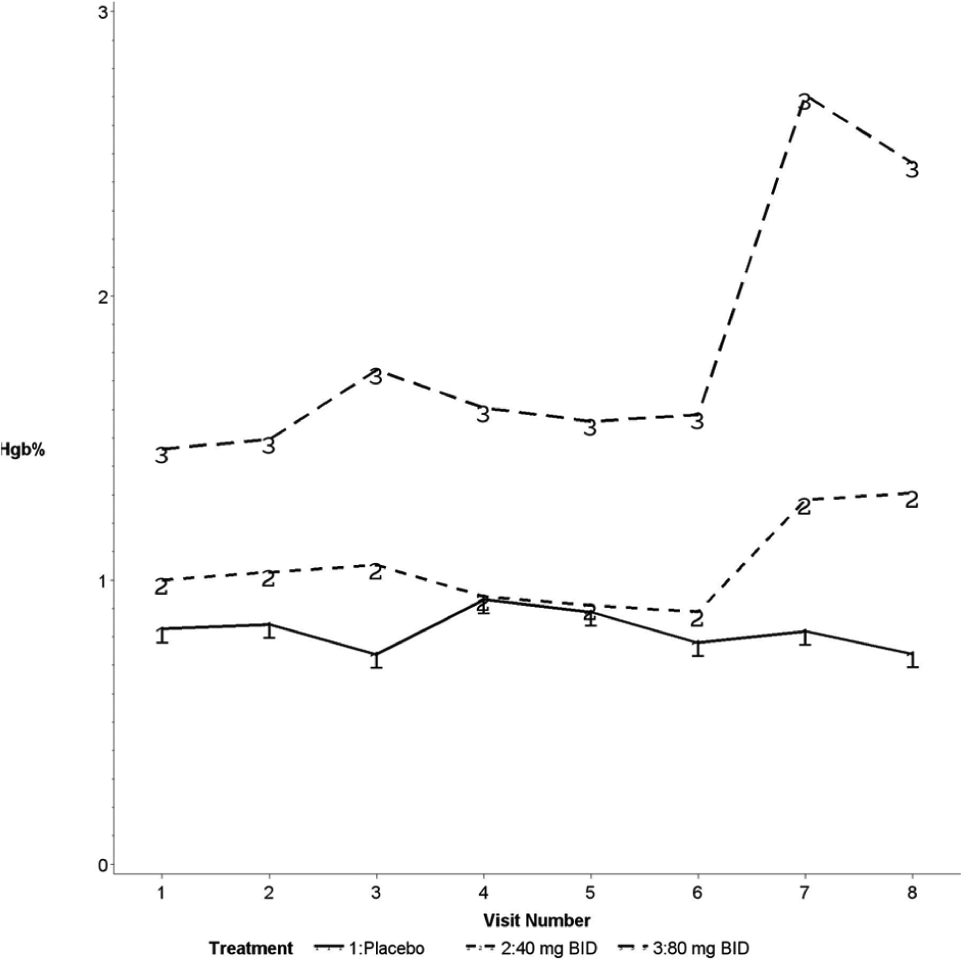
A graph showing the methemoglobin (Hgb%) level at study visits for the three populations. (1 = placebo group; 2 = 40 mg twice-daily group; 3 = 80 mg twice-daily group.)
There was no clinically significant change in the resting heart rate, QT interval, or blood pressure in any of the groups. In addition, orthostatic vital signs were obtained (Appendix 1). The maximum change in orthostatic pulse and blood pressure was seen early after initial drug administration in the 80 mg group and dissipated over the course of the study. For example, at visit 1 post 80 mg dose, the heart rate increased by 6.9 beats/minute and the SBP dropped 4.4 mmHg. However, by visit 6 the postural changes in pulse and blood pressure were numerically similar to placebo. There was no apparent orthostatic change in pulse or blood pressure throughout the study in the 40 mg BID group.
Efficacy data
In the intent-to-treat population there was no significant difference in FMD at baseline among the groups. The difference in FMD from baseline was assessed at 10 weeks (Table 4) and a non-significant trend was noted for worsening of FMD in the placebo and 40 mg BID group whereas maintenance of baseline FMD was observed in the 80 mg BID group (Figure 3). In the diabetic cohort, the change in FMD in those receiving the 80 mg BID dose was significantly greater than those receiving placebo and the 40 mg BID dose (p < 0.01).
Percent flow-mediated dilation of brachial artery.
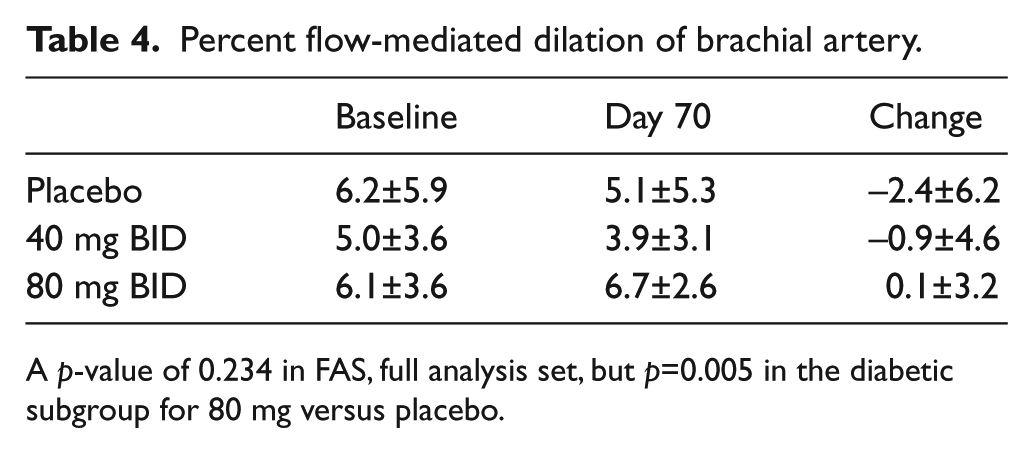
A p-value of 0.234 in FAS, full analysis set, but p=0.005 in the diabetic subgroup for 80 mg versus placebo.
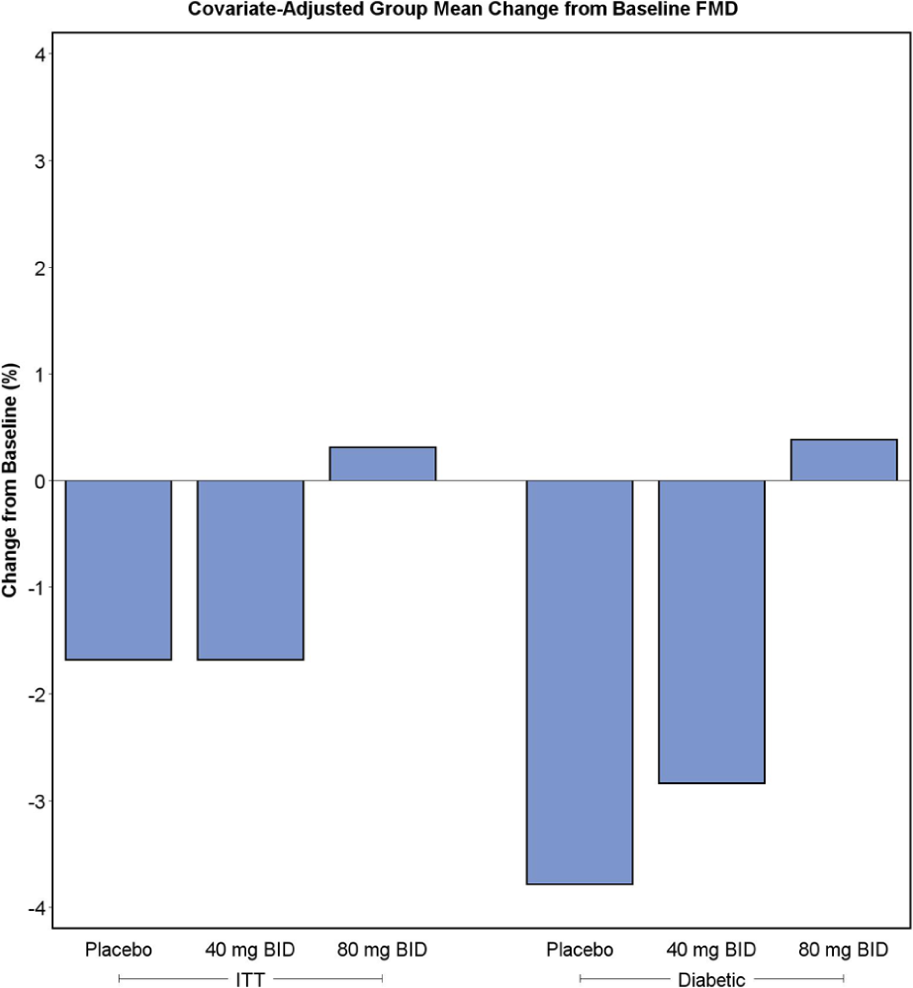
Difference in flow-mediated dilation (FMD) for each population at 10 weeks of study. (ITT, intention-to-treat.)
The 40 mg BID dose group showed a numeric improvement in the 6-minute walk test over time compared to placebo but this was not statistically significant (Figure 4).
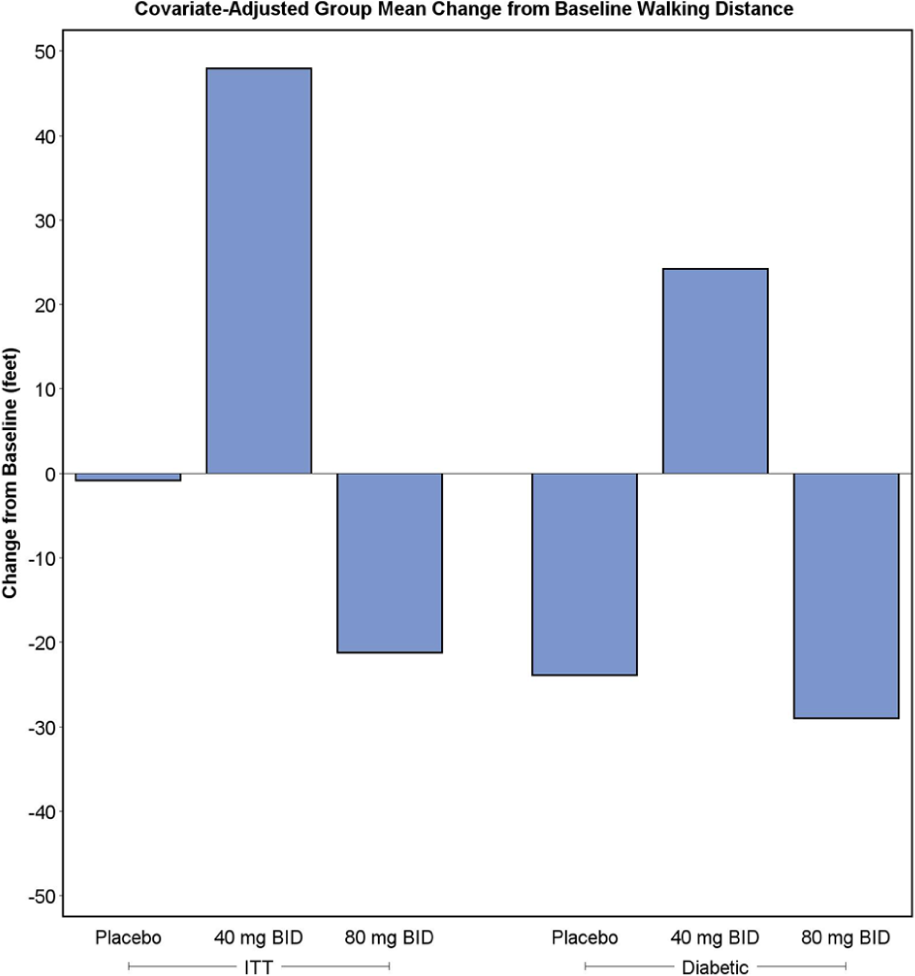
Change in walking distance on 6-minute walk test for each population at 10 weeks of study.
In regards to quality of life, there was no significant overall difference among the groups in composite responses to the RAND 36 questionnaire. However, there was a significant improvement in the pain domain for the physical component of the RAND 36 questionnaire (p<0.05) among patients randomized to the 40 mg group compared with placebo.
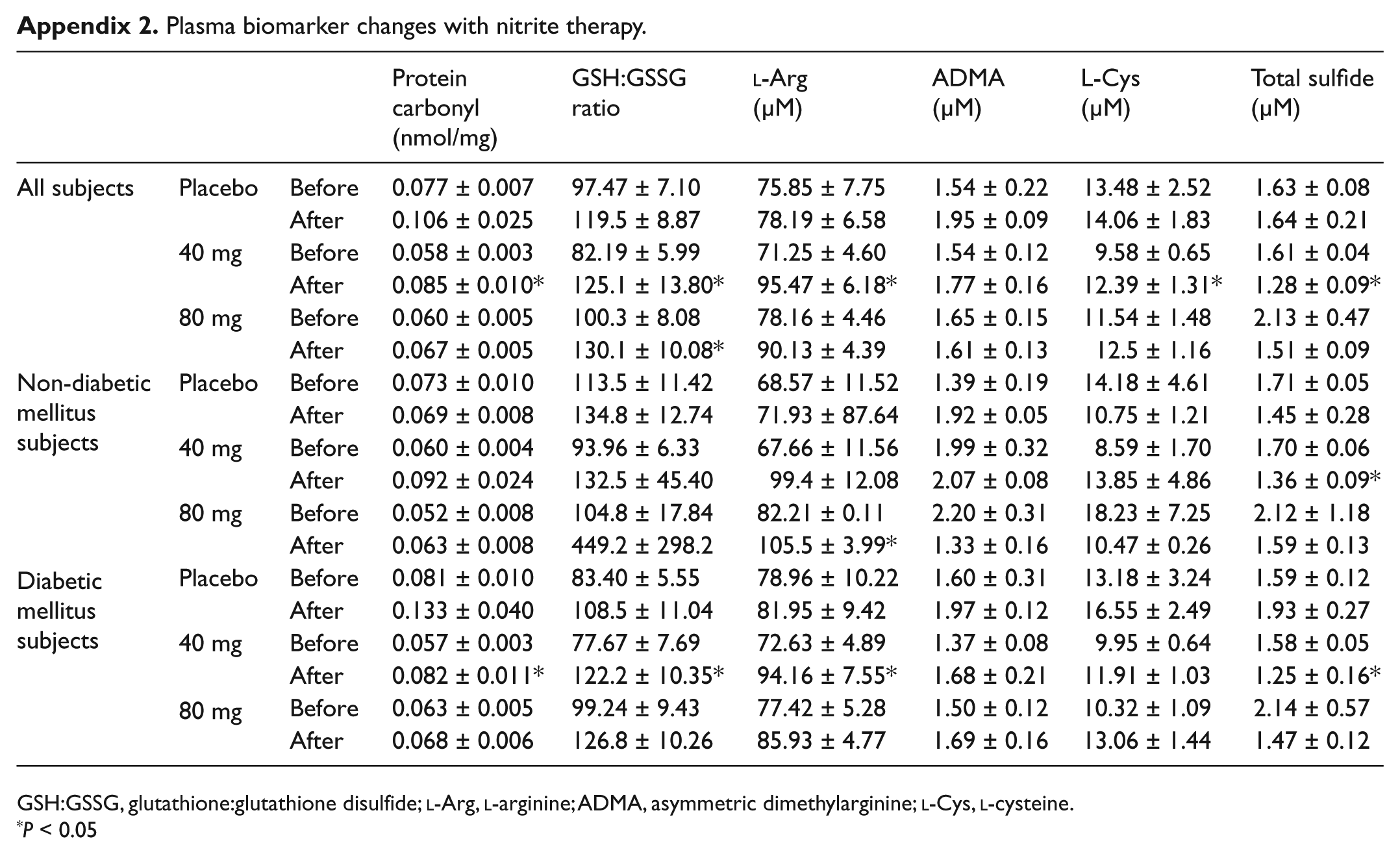
Appendix 2 lists changes in plasma biomarkers before and after sodium nitrite therapy. Plasma protein carbonyl, the GSH:GSSG ratio, and
Discussion
The primary finding of this clinical trial demonstrated that oral sodium nitrite was well tolerated, particularly in the initial randomized dose groups of 40 mg BID and 80 mg BID. Also, patient withdrawals were infrequent and similar across all three groups. However, mild and moderate treatment-emergent adverse events had an apparent dose relationship. This was particularly true at the end of the study when patients initially randomized to the 80 mg dose were escalated to 160 mg BID. In terms of adverse events expected with this drug, there was also a dose–response observed for headaches and dizziness. There was evidence for an acute, but not chronic, postural increase in heart rate and decrease in blood pressure at the 80 mg dose, attesting to the vasodilatory effects of action of nitrite. MetHb production did not increase, except for the last week at the160 mg dose that did not surpass 3% MetHb.
Endothelial function (FMD) studies demonstrated a numeric decrease from baseline for the placebo and 40 mg dose groups. However, the 80 mg dose showed a positive trend for improvement in FMD. These changes in FMD were not statistically significant in the final analysis set, but, in a pre-specified secondary population of patients with diabetes, the 80 mg BID dose was significantly better than 40 mg BID or placebo.
There was no clinically significant increase in the 6-minute walk time, although there was a trend to an improvement with the 40 mg BID dose. The quality of life questionnaires did not demonstrate a significant improvement in most parameters; however, there did appear to be a dose-dependent numeric improvement in the patients’ assessment of overall physical functioning, emotional well-being, pain and social functioning. As with the 6-minute walk test, patients treated with the 40 mg BID dose felt that their general health was improved and reported a statistically significant (p < 0.05) reduction in pain.
Experimental studies have shown that sodium nitrite enhances perfusion of chronically ischemic tissues. Twice-daily intraperitoneal sodium nitrite injections over a range of doses significantly increased the perfusion of murine ischemic hind-limbs secondary to permanent femoral artery ligation.8,20 Sodium nitrite stimulates increased vascular density in chronically ischemic tissues, possibly by increasing endothelial cell proliferation and stimulation of collateral vessel remodeling.8,21 Because patients with diabetes and PAD, either in combination or alone, have endothelial dysfunction, decreased NO bioavailability, and depletion of NO stores, sodium nitrite may improve endothelial function. Nitrite anion acts as a NO reservoir that can be readily converted to NO by a non-enzymatic one electron reduction by deoxyhemoglobin, deoxymyoglobin, xanthine oxidoreductase and other mechanisms, making it a unique candidate for potential therapeutic effect in ischemic tissues.22,23 In this way, nitrite reduction to NO preferentially occurs during tissue hypoxia where O2 concentrations for NOS function (O2 Km = 24 µM) may be compromised. Thus, nitrite therapy could be a useful alternative source of bioavailable NO to alleviate endothelial cell dysfunction.
There are several drugs that demonstrated improvement in endothelial function including HMG-CoA reductase inhibitors,24–26 erythropoietin (and analogues), 27 and modulators of the renin-angiotensin system.28–30 The data derived from this study indicate that oral administration of sodium nitrite in the doses administered is safe and the 80 mg BID dose may afford protection against worsening endothelial function among patients with PAD and diabetes.
In conclusion, therapy with sodium nitrite appears safe in patients with PAD. Although the primary efficacy parameter of FMD in the entire study population failed to show a statistically significant effect, those with diabetes who were the original population targeted in the study had a significant improvement in endothelial dysfunction. There was a trend toward an improvement in the functional parameters; however, the study was not powered to fully evaluate these endpoints. Thus, this study demonstrates that sodium nitrite was well tolerated at the 40 and 80 mg BID doses and had signals of biologic activity and possibly clinical benefit that should be explored in a larger and fully powered trial.
Footnotes
Appendix
Plasma biomarker changes with nitrite therapy.
| Protein carbonyl |
GSH:GSSG ratio | ADMA |
L-Cys |
Total sulfide |
||||
|---|---|---|---|---|---|---|---|---|
| All subjects | Placebo | Before | 0.077 ± 0.007 | 97.47 ± 7.10 | 75.85 ± 7.75 | 1.54 ± 0.22 | 13.48 ± 2.52 | 1.63 ± 0.08 |
| After | 0.106 ± 0.025 | 119.5 ± 8.87 | 78.19 ± 6.58 | 1.95 ± 0.09 | 14.06 ± 1.83 | 1.64 ± 0.21 | ||
| 40 mg | Before | 0.058 ± 0.003 | 82.19 ± 5.99 | 71.25 ± 4.60 | 1.54 ± 0.12 | 9.58 ± 0.65 | 1.61 ± 0.04 | |
| After | 0.085 ± 0.010* | 125.1 ± 13.80* | 95.47 ± 6.18* | 1.77 ± 0.16 | 12.39 ± 1.31* | 1.28 ± 0.09* | ||
| 80 mg | Before | 0.060 ± 0.005 | 100.3 ± 8.08 | 78.16 ± 4.46 | 1.65 ± 0.15 | 11.54 ± 1.48 | 2.13 ± 0.47 | |
| After | 0.067 ± 0.005 | 130.1 ± 10.08* | 90.13 ± 4.39 | 1.61 ± 0.13 | 12.5 ± 1.16 | 1.51 ± 0.09 | ||
| Non-diabetic mellitus subjects | Placebo | Before | 0.073 ± 0.010 | 113.5 ± 11.42 | 68.57 ± 11.52 | 1.39 ± 0.19 | 14.18 ± 4.61 | 1.71 ± 0.05 |
| After | 0.069 ± 0.008 | 134.8 ± 12.74 | 71.93 ± 87.64 | 1.92 ± 0.05 | 10.75 ± 1.21 | 1.45 ± 0.28 | ||
| 40 mg | Before | 0.060 ± 0.004 | 93.96 ± 6.33 | 67.66 ± 11.56 | 1.99 ± 0.32 | 8.59 ± 1.70 | 1.70 ± 0.06 | |
| After | 0.092 ± 0.024 | 132.5 ± 45.40 | 99.4 ± 12.08 | 2.07 ± 0.08 | 13.85 ± 4.86 | 1.36 ± 0.09* | ||
| 80 mg | Before | 0.052 ± 0.008 | 104.8 ± 17.84 | 82.21 ± 0.11 | 2.20 ± 0.31 | 18.23 ± 7.25 | 2.12 ± 1.18 | |
| After | 0.063 ± 0.008 | 449.2 ± 298.2 | 105.5 ± 3.99* | 1.33 ± 0.16 | 10.47 ± 0.26 | 1.59 ± 0.13 | ||
| Diabetic mellitus subjects | Placebo | Before | 0.081 ± 0.010 | 83.40 ± 5.55 | 78.96 ± 10.22 | 1.60 ± 0.31 | 13.18 ± 3.24 | 1.59 ± 0.12 |
| After | 0.133 ± 0.040 | 108.5 ± 11.04 | 81.95 ± 9.42 | 1.97 ± 0.12 | 16.55 ± 2.49 | 1.93 ± 0.27 | ||
| 40 mg | Before | 0.057 ± 0.003 | 77.67 ± 7.69 | 72.63 ± 4.89 | 1.37 ± 0.08 | 9.95 ± 0.64 | 1.58 ± 0.05 | |
| After | 0.082 ± 0.011* | 122.2 ± 10.35* | 94.16 ± 7.55* | 1.68 ± 0.21 | 11.91 ± 1.03 | 1.25 ± 0.16* | ||
| 80 mg | Before | 0.063 ± 0.005 | 99.24 ± 9.43 | 77.42 ± 5.28 | 1.50 ± 0.12 | 10.32 ± 1.09 | 2.14 ± 0.57 | |
| After | 0.068 ± 0.006 | 126.8 ± 10.26 | 85.93 ± 4.77 | 1.69 ± 0.16 | 13.06 ± 1.44 | 1.47 ± 0.12 |
GSH:GSSG, glutathione:glutathione disulfide;
P < 0.05
Acknowledgements
We thank Elizabeth Medenilla, MD, Chandra Sehgal, PhD and Susan Schultz, all at the Perelman School of Medicine at the University of Pennsylvania School of Medicine, for their assistance. Dr Mohler is the guarantor of this work and, as such, had full access to all the data in the study and takes responsibility for the integrity of the data and the accuracy of the data analysis.
Declaration of conflicting interest
In regards to potential conflict of interest, William Hiatt received funding from TheraVasc to conduct the study at CPC Clinical Research, a non-profit clinical research organization affiliate of the University of Colorado. Brian H Annex received honorarium and equity shares from TheraVasc and served as a Medical Advisor and Steering Committee member. Christopher G Kevil has IP interest in nitrite therapy for cardiovascular disease, has ownership interest, and is Chief Scientific Officer of TheraVasc. None of the other authors report a potential conflict of interest.
Funding
TheraVasc, Inc., Cleveland, OH funded the entire study.