
Abstract
Dimethylarginine dimethyl-aminohydrolase 1 (DDAH1) is a metabolic enzyme for asymmetric dimethylarginine (ADMA), both of which are closely related to endothelial function. Endothelial dysfunction, a main risk factor of cardiovascular diseases, can be attributed to insulin resistance. We aimed to determine the effects of atorvastatin, an endothelium-protective drug, on DDAH1/ADMA in insulin-resistant rats. Insulin resistance in male Sprague-Dawley rats was induced with a high-fat diet for 8 weeks. Some rats received atorvastatin (30 mg/kg/day) for an additional 8 weeks. Insulin-resistant rats exhibited not only decreases in the DDAH activity and aortic expression of DDAH1 and sterol regulatory element-binding protein 1 (SREBP1) but also increases in plasma ADMA levels, all of which were inhibited by atorvastatin. Insulin sensitivity and DDAH activity showed a significant positive correlation. In conclusion, our results suggest that atorvastatin may modulate DDAH1/ADMA to improve endothelial function in insulin-resistant rats; SREBP1 may also play a role in this.
Introduction
Insulin-resistance, prevalent among people with a high-fructose diet, is a multifactorial disorder with decreased sensitivity and responsiveness to insulin metabolic actions that promote glucose disposal. 1 Endothelial dysfunction, also characterized by insulin resistance, is a major risk factor of cardiovascular diseases. It is well understood that insulin resistance and endothelial dysfunction are linked not only by common pathogenetic mechanisms that involve deranged insulin signaling pathways but also by other indirect mechanisms that control hormone functions.2,3
Asymmetric dimethylarginine (ADMA), an endogenous inhibitor of nitric oxide synthase (NOS), plays an important role in endothelial function and has been considered as a biomarker for major cardiovascular events and mortality in cohorts with high, intermediate, and low overall cardiovascular risks. 4 ADMA is metabolized by the enzyme named dimethylarginine dimethyl-aminohydrolase (DDAH). Two DDAH isoforms, DDAH1 and DDAH2, 5 share a similar enzymatic activity but have distinct tissue distributions. However, it is clear that both isoforms are expressed in the same cell types in cardiovascular tissues. 6 Physiological studies have reported that DDAH inhibition affected ADMA levels and NO biosynthesis in isolated blood vessels. 7 Therefore, it is believed that DDAH indirectly regulates NO production by modulating ADMA in vivo. 8 Moreover, heterozygous deletion studies suggest that DDAH1 not only is the primary isoform responsible for metabolizing ADMA 9 but also plays a role in endothelial function and cardiovascular hemodynamics. 10 Furthermore, DDAH overexpression has also been reported to enhance insulin sensitivity. 11 However, the regulation of DDAH by insulin resistance is still poorly understood. In this study, we hypothesize that endothelial dysfunction induced by insulin resistance may result from the reduced DDAH activity.
Sterol regulatory element-binding proteins (SREBPs) are transcription factors that regulate the expression of genes involved in lipid homeostasis and glucose metabolism. Two distinct SREBPs, SREBP1 and SREBP2, have been identified, and their expression, transport, and processing can be regulated by sterols. SREBP1 has also been shown to be regulated by insulin.12–14 However, these previous studies mainly focused on SREBP1 in hepatic cells. Based on the fact that the DDAH1 promoter contains sterol regulatory elements, we hypothesize that SREBP1 may play the role of a mediator in the reduction of DDAH activity induced by insulin resistance. Here, we investigate the effect of insulin resistance on SREBP1 expression in endothelial cells as well as the potential role for SREBP1 in endothelial function.
Furthermore, atorvastatin, an inhibitor of 3-hydroxy-3-methyl glutaryl coenzyme A (HMG-CoA) reductase, is proved to have protective effects on endothelial function. The compound was also reported to regulate SREBP1 in the process of lipid lowering, 15 glucose metabolism, and insulin resistance. 16 However, no study has examined the effect of atorvastatin on DDAH activity in vitro or in vivo.
Therefore, the purpose of this study was to investigate the effects of insulin resistance and atorvastatin on the DDAH1/ADMA system in rats with endothelial dysfunction.
Methods
Animal care
Four-week-old male Sprague-Dawley (SD) rats provided by the Animal Center of Central South University (China) were housed at a constant temperature (25°C) with a 12-hour light/dark cycle. Animal care was in compliance with the Guidelines for the Care and Use of Laboratory Animals.
Animal treatment
Eight-week-old SD rats (weighing 200–250 g) were randomly divided into two groups: (1) the control group (CON; n = 10), in which rats were fed with standard rodent chow and water ad libitum (protein, 20 kcal%; carbohydrate, 70 kcal%; and lipid, 10 kcal%) and (2) the high-fat diet group (HFD; n = 20), in which rats were fed with fat-rich chow and water ad libitum (protein, 20 kcal%; carbohydrate, 35 kcal%; and lipid, 45 kcal%, predominantly in the form of lard). At the end of week 8, fasting plasma glucose and insulin levels were measured, and insulin sensitivity was evaluated by calculating the homeostatic model assessment-insulin resistance (HOMA-IR) index as [(fasting insulin, mU/ml) × (fasting glucose, mmol/L)/22.5]. 17 Studies have previously demonstrated that a high-fat diet induced insulin resistance in rats.18–21 Next, the HFD group was further divided into two groups. The first group (IR+A; n = 10) received atorvastatin (30 mg/kg/day; Pfizer Pharmaceuticals) and the second group (IR; n = 10) was kept on vehicle (water) for an additional 8 weeks. Body weights were measured weekly. Fasting glucose and insulin levels were measured at the beginning of week 8 and week 16. Finally, the aorta and plasma were collected and stored at −80°C for further studies.
Insulin sensitivity assessment and insulin resistance test
Rats were fasted for 12 hours before blood samples were obtained and measured for fasting plasma glucose and insulin levels. The HOMA-IR index was calculated to evaluate insulin sensitivity, as described before.
At the end of the experiment, an insulin resistance test was performed. Fasted rats were given 0.4 U of insulin intraperitoneally, and plasma glucose levels were measured at 0, 40, and 90 minutes.
Biochemical analysis
The total concentration of nitrite and nitrate was measured to reflect plasma NO levels. Absorbance was determined at 540 nm with a spectrophotometer. NOS activity was determined by measuring the conversion of 3H-
Plasma ADMA concentrations and aortic DDAH activity
Plasma ADMA concentrations were measured by high-performance liquid chromatography using precolumn derivatization with o-phthaldialdehyde, as described previously.23,24 Aortic DDAH activity was measured by determining the
Determination of endothelium-dependent vasodilator responses
Acetylcholine (ACh)-induced endothelium-dependent relaxation of thoracic aortas was determined as described previously.23,26,27
Western immunoblotting
Frozen aortic tissues were rinsed in cold PBS and crushed in liquid nitrogen. Tissue samples were homogenized in lysis buffer, and protein concentrations in the supernatant were determined with the Bradford assay. Equal amounts of proteins were subjected to 8% sodium dodecyl sulfate-polyacrylamide gel electrophoresis and transferred to polyvinylidene difluoride membranes. Membranes were then probed with primary antibodies against SREBP1 and DDAH1 (Santa Cruz Biotechnology) at 4°C overnight, followed by incubation with the secondary antibody (Santa Cruz) for 2 hours at room temperature. Bound antibody complexes were subsequently detected using an electrochemiluminescence reagent. Expression levels of SREBP1 and DDAH1 were normalized to that of glyceraldehyde-3-phosphate dehydrogenase (GAPDH).
Real-time PCR
Total RNA was isolated from rat thoracic aorta with TRIzol reagent (Invitrogen). First-strand cDNA was synthesized from 5 μg of total RNA with random hexamer primers and Superscript II Reverse Transcriptase (Invitrogen). GAPDH was used as a control for all reactions. Primer sequences are as follows: DDAH1 (forward, 5′–AGCCACCCCTCGGTC TTCGG–3′; reverse, 5′–GTCTCCTCGCACACCACGGC–3′), SREBP1 (forward, 5′–CAGGCAGGCCCCTTGCAGAC–3′; reverse, 5′–GTGCGCTTCTCACCACGGCT–3′), and GAPDH (forward, 5′–CTCATGACCACAGTCCATGC–3′; reverse, 5′–TTCAGCTCTGGGATGACCTT–3′). mRNA expression levels of SREBP1 and DDAH1 were normalized to the GAPDH control.
Statistical analysis
All data were presented as mean ± standard error (SEM). Comparisons between two groups were assessed by Student’s paired t-test. Comparisons between three or more groups were performed using one-way analysis of variance (ANOVA). Pearson correlation coefficients were calculated when indicated. All the values of p < 0.05 were considered statistically significant. For statistical analysis, the SPSS 17.0 software was used.
Results
Body weights and biochemical characteristics
As shown in Table 1, there were no significant differences in body weights, plasma total cholesterol, or LDL-C and HDL-C levels among the control, insulin-resistant, and atorvastatin-treated rats. In contrast, higher levels of plasma triglycerides and CRP were observed in insulin-resistant rats than in control rats (1.45 ± 0.41 vs 0.9 ± 0.24 mmol/L, p < 0.05; 0.3 ± 0.1 vs 0.12 ± 0.1 mg/L, p < 0.01; respectively). Significantly, atorvastatin treatment reduced both plasma triglycerides and CRP levels in insulin-resistant rats (0.82 ± 0.3 mmol/L and 0.03 ± 0.01 mg/L, respectively; p < 0.01).
Body weights and biochemical characteristics among different groups
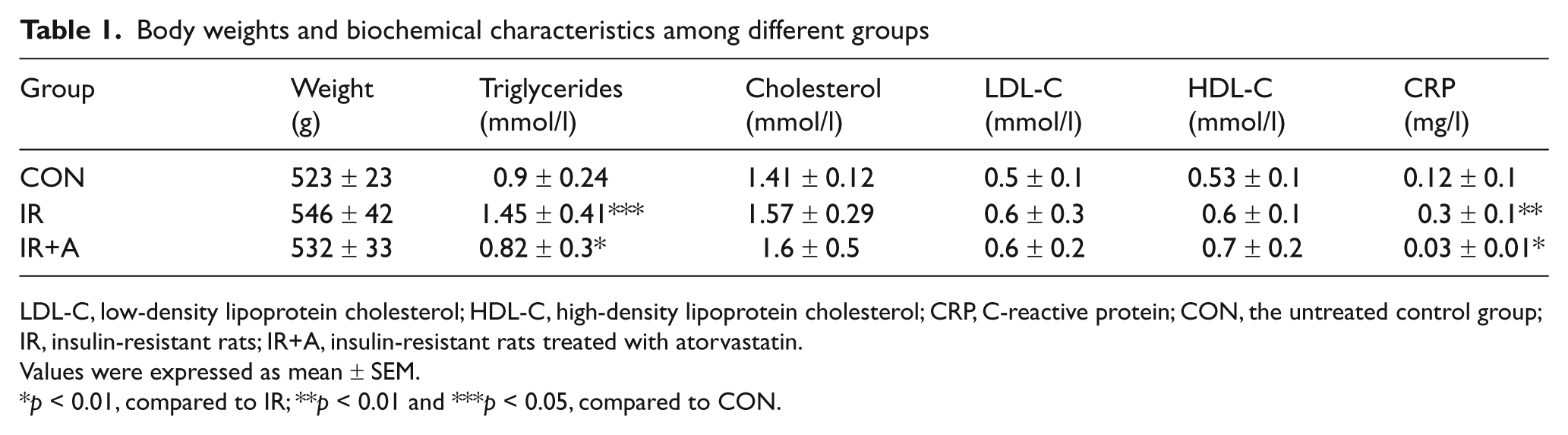
LDL-C, low-density lipoprotein cholesterol; HDL-C, high-density lipoprotein cholesterol; CRP, C-reactive protein; CON, the untreated control group; IR, insulin-resistant rats; IR+A, insulin-resistant rats treated with atorvastatin.
Values were expressed as mean ± SEM.
p < 0.01, compared to IR; **p < 0.01 and ***p < 0.05, compared to CON.
Insulin sensitivity and insulin tolerance
Insulin sensitivity was evaluated by calculating the HOMA-IR index. The HFD rats showed a significantly higher HOMA-IR than the control group (3.06 ± 0.6 vs 1.89 ± 0.3, p < 0.05), and such high insulin sensitivity in insulin-resistant rats was reduced by atorvastatin treatment (3.06 ± 0.6 vs 2.38 ± 0.5, p < 0.05) (Figure 1a).
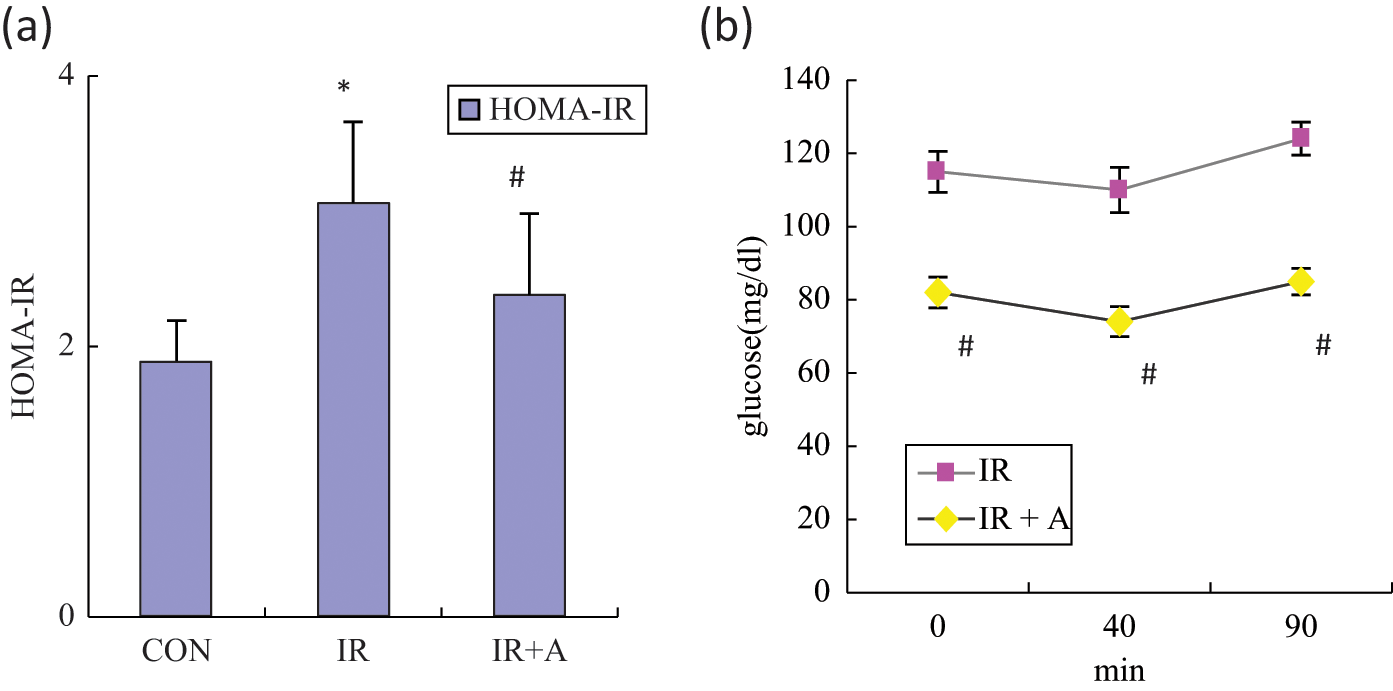
The effects of atorvastatin on insulin tolerance in insulin-resistant rats. An insulin tolerance test was performed after the administration of atorvastatin (30 mg/kg/day for 8 weeks, n = 10). (a) HOMA-IR. (b) Mean blood glucose. CON, the untreated control group; IR, insulin-resistant rats; IR+A, insulin-resistant rats treated with atorvastatin. Values expressed as mean ± SEM. *p < 0.05, compared to CON; #p < 0.05, compared to IR.
By using the insulin tolerance test, blood glucose levels in atorvastatin-treated insulin-resistant rats were also significantly lower than those in the untreated group at all times after insulin injection (Figure 1b).
Effects of insulin resistance and atorvastatin on plasma ADMA levels and aortic DDAH activity
Plasma ADMA concentrations were significantly increased in insulin-resistant rats compared to controls (1.1 ± 0.24 vs 0.52 ± 0.1 μmol/L, p < 0.01), whereas aortic DDAH activity was reduced (0.05 ± 0.01 vs 0.12 ± 0.01 U/g protein, p < 0.01) (Figure 2a and b). Further atorvastatin treatment of insulin-resistant rats not only significantly decreased plasma ADMA levels (0.87 ± 0.22 vs 1.1 ± 0.24 μmol/L, p < 0.05) but also enhanced aortic DDAH activity by 18% (0.07 ± 0.012 vs 0.05 ± 0.01 U/g protein, p < 0.05) (Figure 2a and b).
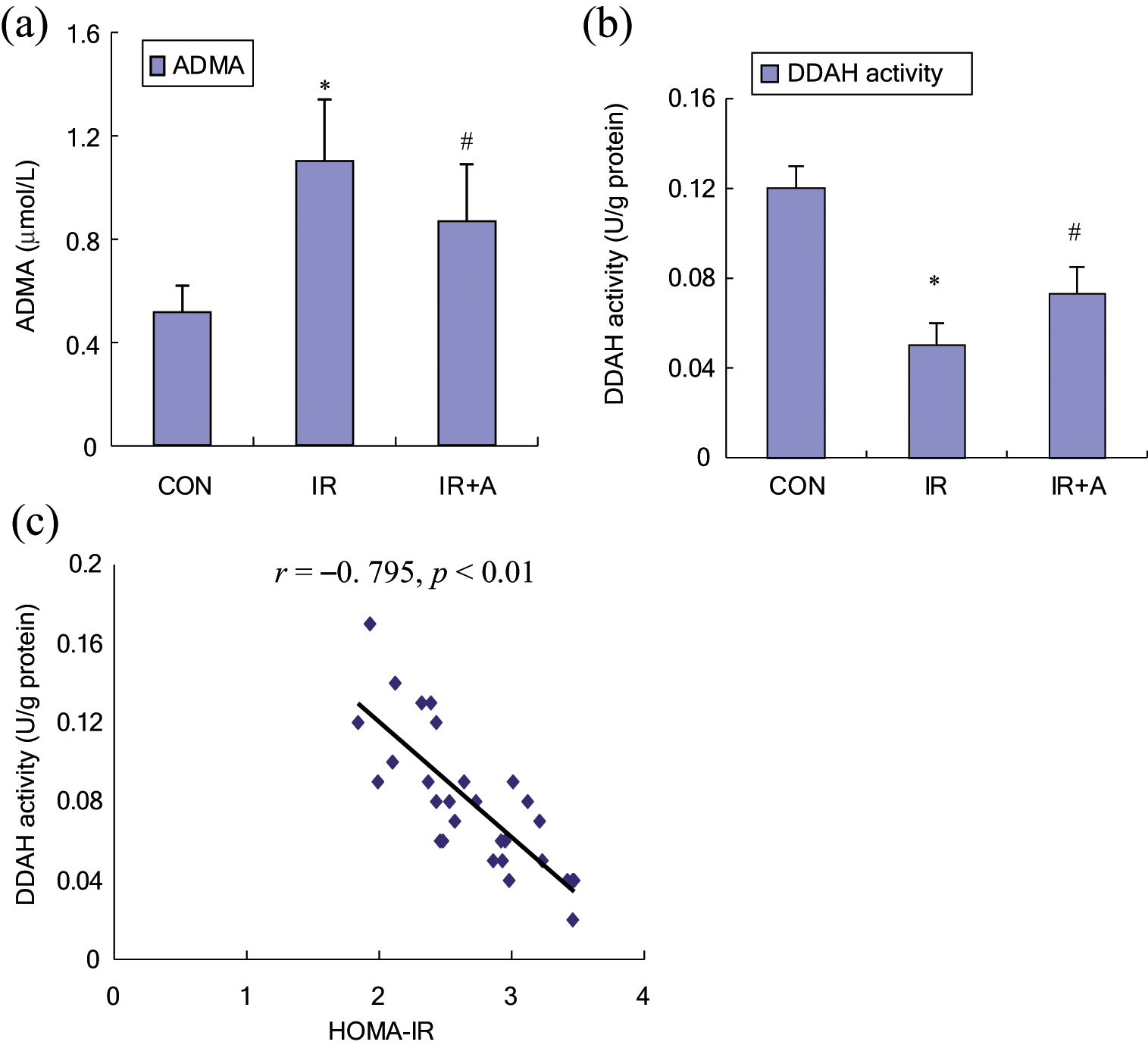
The effects of insulin resistance and atorvastatin on plasma ADMA levels (a) and vascular DDAH activities (b). CON, the untreated control group; IR, insulin-resistant rats; IR+A, insulin-resistant rats treated with atorvastatin. Values expressed as mean ± SEM, n = 10. *p < 0.01, compared to CON; #p < 0.05, compared to IR. (c) Significant negative correlation between HOMA-IR and aortic DDAH activity in the IR+A group, indicating a positive correlation between insulin sensitivity and DDAH activity.
As shown in Figure 2c, a significant negative correlation between HOMA-IR and aortic DDAH activity (r = −0.795, p < 0.01) was observed, which in turn indicates a positive correlation between insulin sensitivity and aortic DDAH activity.
Effects of insulin resistance and atorvastatin on plasma NO concentrations and NOS activity
Insulin-resistant rats showed decreased plasma NO levels and NOS activity compared to the control group (25.6 ± 7.4 vs 62.4 ± 4.9 μmol/L, 19 ± 3.2 vs 35 ± 4.8 U/ml, respectively; p < 0.01). Significantly, atorvastatin treatment of insulin-resistant rats was able to increase the plasma NO levels (43.53 ± 8.2 vs 25.6 ± 7.4 μmol/L, p < 0.01) and NOS activity (26 ± 3.5 vs 19 ± 3.2 U/ml, p < 0.05) (Figure 3a and b).
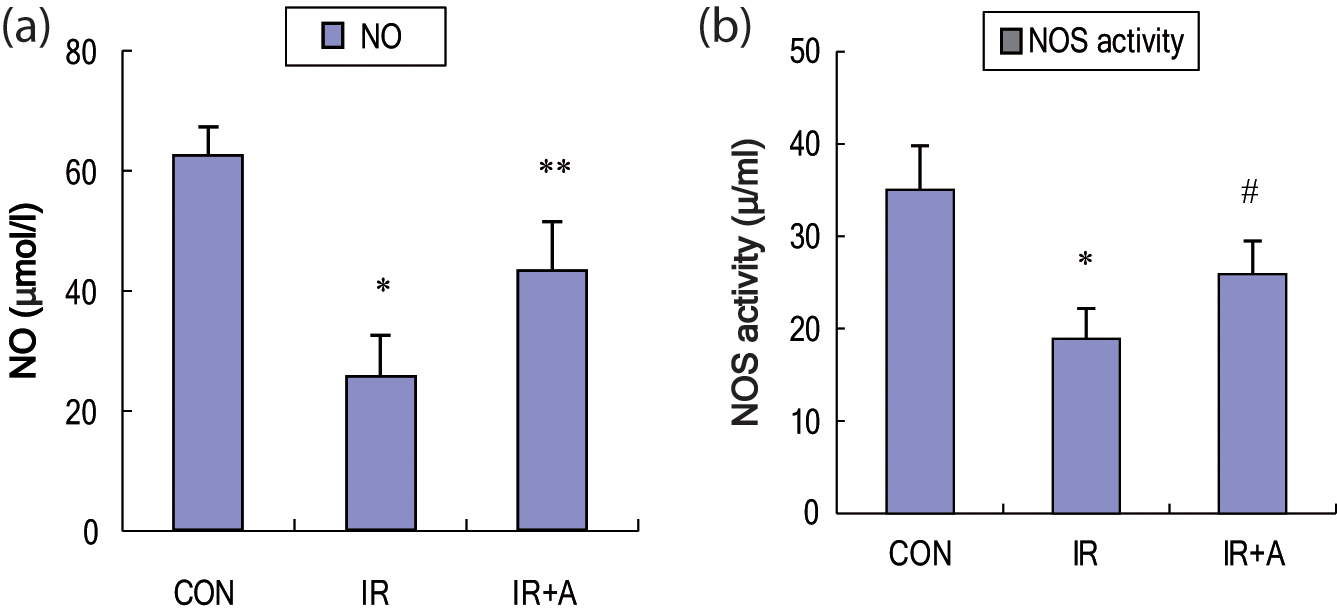
The effects of insulin resistance and atorvastatin on plasma NO levels (a) and NOS activities (b). CON, the untreated control group; IR, insulin-resistant rats; IR+A, insulin-resistant rats treated with atorvastatin. Values expressed as mean ± SEM, n = 10. *p < 0.01, compared to CON; **p < 0.01 and #p < 0.05, compared to IR.
Effects of insulin resistance and atorvastatin on acetylcholine-induced vasodilator responses of thoracic aorta
Acetylcholine (ACh) is noted to cause endothelium-dependent relaxation in a dose-dependent manner. Insulin resistance in HFD rats significantly attenuated ACh-induced endothelium-dependent relaxation; in contrast, atorvastatin treatment preserved the control-level relaxation (Figure 4).
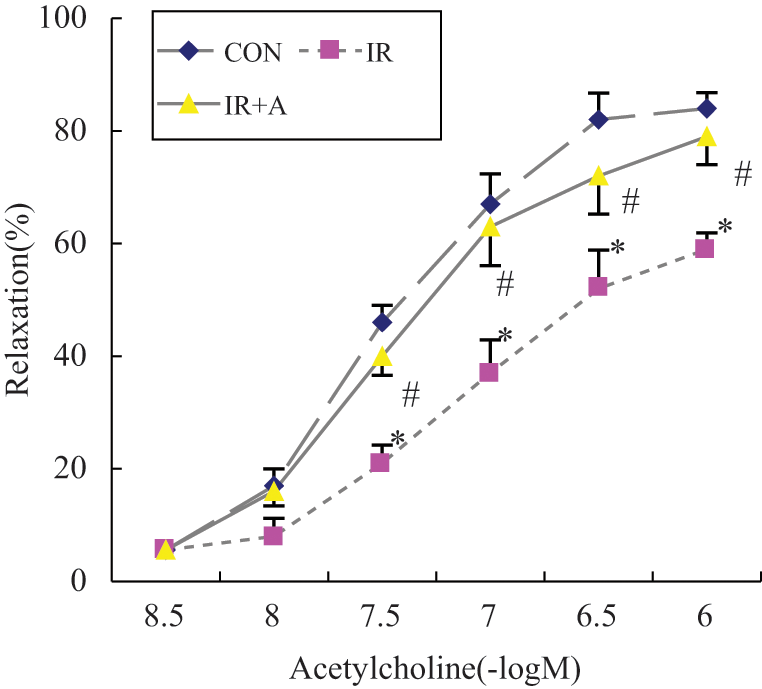
The effects of insulin resistance and atorvastatin on acetylcholine (ACh)-induced thoracic aorta endothelium relaxation. CON, the untreated control group; IR, insulin-resistant rats; IR+A, insulin-resistant rats treated with atorvastatin. Values expressed as mean ± SEM. *p < 0.01, compared to CON; #p < 0.01, compared to IR.
Effects of insulin resistance and atorvastatin on SREBP1 and DDAH1 expression in thoracic aorta
The mRNA and protein expression of SREBP1 and DDAH1 in the thoracic aorta of insulin-resistant rats were significantly lower than those in controls; however, all levels were restored by further atorvastatin treatment (Figure 5).
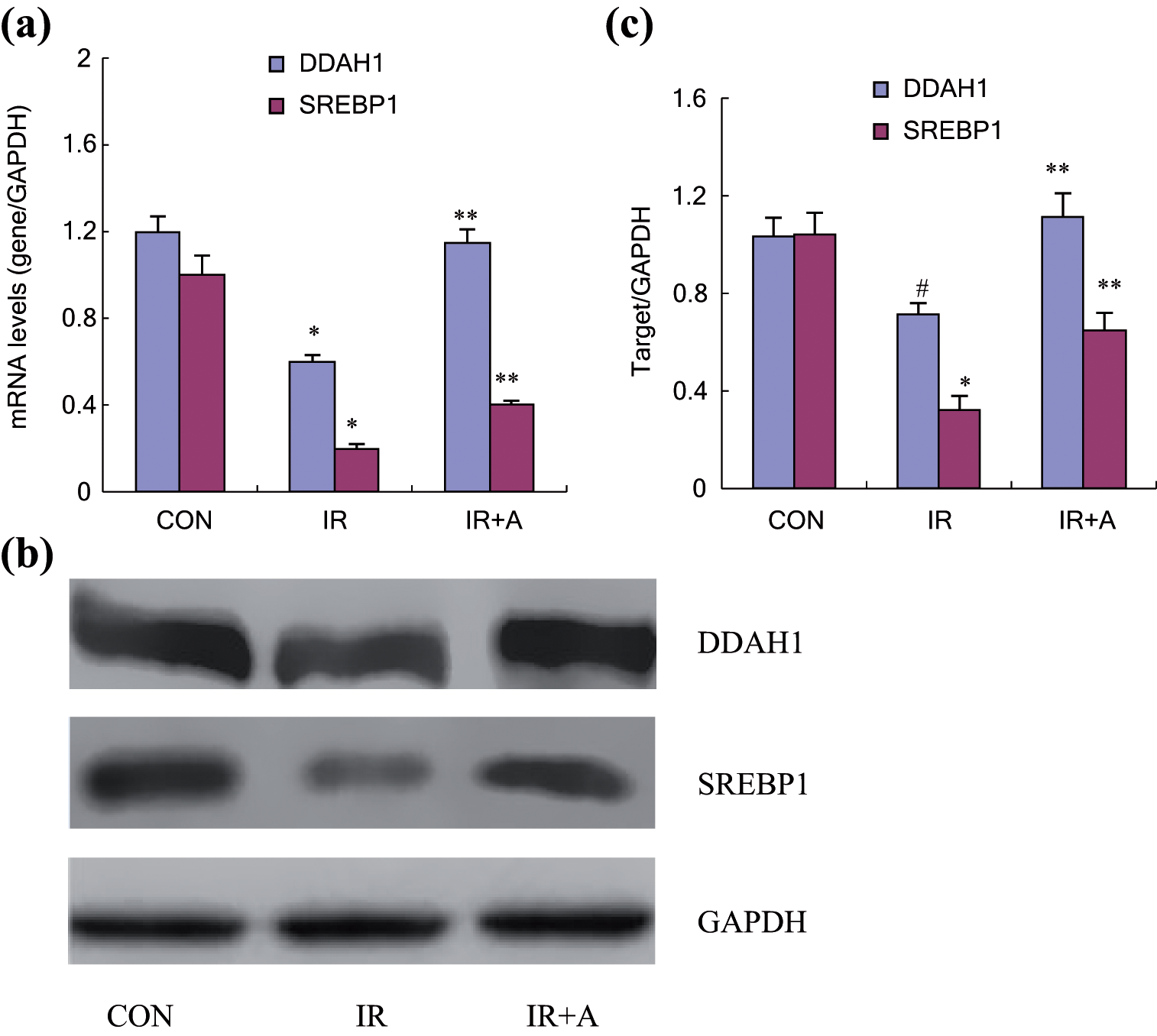
The effects of insulin resistance and atorvastatin on expression levels of SREBP1 and DDAH1 mRNA (a) and proteins (b and c) in thoracic aorta. (a) mRNA levels were analyzed by quantitative RT-PCR. (b and c) Protein levels were determined by western blot analysis and densitometry. The GAPDH expression was used as a control. CON, the untreated control group; IR, insulin-resistant rats; IR+A, insulin-resistant rats treated with atorvastatin. *p < 0.01 and #p < 0.05, compared to CON; **p < 0.01, compared to IR.
Discussion
Insulin resistance plays an important role in the development of endothelial dysfunction and cardiovascular diseases. ADMA has been associated with both insulin resistance and endothelial dysfunction.28–33 Specifically, inhibition of ADMA was shown to provide a protective effect on endothelial function. DDAH1 is recognized as the dominant isoform metabolizing ADMA, and impaired DDAH activity can lead to increases in plasma ADMA concentrations. 34 To date, the effects of insulin resistance and atorvastatin on DDAH1 and ADMA remain unclear. In this current study, we provide the first evidence that aortic DDAH1 expression and DDAH activity were decreased in insulin-resistant rats, which is in agreement with previous reports that DDAH transgenic mice were sensitive to insulin. 11 We also showed that atorvastatin treatment can restore DDAH activity and in turn decrease plasma ADMA levels.
In this study, we successfully established an insulin-resistant animal model by feeding rats with a high-fat diet for 8 weeks, which was confirmed by calculating the HOMA-IR index. The plasma triglyceride levels in insulin-resistant rats were higher than those in the control animals, while the total weights, cholesterol, LDL, or HDL showed no significant differences. Further atorvastatin treatment of insulin-resistant rats was able to improve insulin sensitivity and lower plasma triglyceride levels.
Atorvastatin has been considered as an endothelium-protective drug in metabolic and cardiovascular diseases.35–37 However, its molecular mechanism still needs further investigation. ADMA, produced mainly in endothelial cells, is an endogenous NOS inhibitor, and higher ADMA levels have been reported under various insulin-resistant conditions.33,38,39 The effect of atorvastatin on plasma ADMA remains controversial. Paiva et al. first reported that atorvastatin treatment had no clear influence on plasma ADMA, 40 which was further supported by later studies.41,42 However, a study recently showed that atorvastatin decreased serum ADMA in ischemic stroke patients, 43 which is consistent with our results that atorvastatin treatment significantly improved insulin sensitivity and decreased plasma ADMA concentrations in insulin-resistant rats. Therefore, our findings imply that ADMA may be a mediator through which atorvastatin modulates endothelial function.
To further investigate the mechanism for the ADMA reduction after atorvastatin treatment, we examined DDAH1 expression and DDAH activity in aorta. Several studies have provided evidence that DDAH1 regulates ADMA concentrations and endothelial function in vivo.7,10 DDAH1 overexpression in transgenic mice was also shown to result in a twofold reduction in plasma ADMA levels and a twofold increase in tissue NOS activity.
44
However, the effect of statins on DDAH activity was less studied in the past. Yin and Xiong reported that pravastatin was able to restore DDAH activity in aorta after exposure to glycated protein.
45
In this study, we demonstrate a reduction of aortic DDAH1 expression and DDAH activity in insulin-resistant rats and show that atorvastatin attenuated this reduction and improved insulin sensitivity. Moreover, a positive correlation was also observed between insulin sensitivity and aortic DDAH activity. In summary, we observed that atorvastatin treatment not only resulted in a 21% reduction in plasma ADMA concentrations in insulin-resistant rats but also restored the decreased NO levels and NOS activity by 42% and 27%, respectively. The endothelium-dependent relaxation was also improved by atorvastatin. There are two known catabolic pathways for ADMA: renal excretion and enzymatic degradation by DDAH. The latter accounts for > 80% of the ADMA elimination in vivo.
46
DDAH1 gene silencing has been associated with a 48% reduction of
Oxidative LDL has also been reported to increase ADMA levels by modulating the expression of type-1 protein arginine methyltransferases (PRMTs).50,51 However, Ivashchenko CY et al reported that neither simvastatin treatment nor SREBP levels had significant effects on the expression of PRMTs. 52 Further studies are necessary to investigate the possibility that atorvastatin may inhibit oxidized LDL-induced ADMA.
SREBP1 is a subtype of SREBP that can be affected by statins to regulate lipid homeostasis and glucose metabolism. Atorvastatin has been shown to activate SREBP1 in hepatic 15 and human smooth muscle cells. 53 Although the relationship between SREBP1 and DDAH1 is still not concrete, it has been reported that the DDAH1 promoter region contains recognition sites for SREBP binding. Chromatin immunoprecipitation and siRNA studies have also demonstrated the role of SREBPs in DDAH1 activation. 52 Therefore, we hypothesized that SREBP1 may mediate the DDAH1/ADMA regulation by atorvastatin. We examined the expression of both SREBP1 and DDAH1 in thoracic aorta. Our results show that their mRNA and protein expression were significantly downregulated in insulin-resistant rats, and atorvastatin treatment was able to significantly restore all the levels. The change of SREBP1 in thoracic aorta is consistent with the change of DDAH1. A further in vitro study is underway to address the correlation between SREBP1 and DDAH1.
In this study, we have not yet clarified the molecular mechanisms for the upregulated DDAH1 expression and DDAH activity after atorvastatin treatment. However, based on a previous study, 52 we can hypothesize that SREBP1 may be involved in the DDAH1 regulation. To test this hypothesis, inhibition and overexpression of SREBP1 need to be introduced in in vitro studies.
In conclusion, high-fat diet-induced insulin resistance was able to not only downregulate aortic DDAH1 expression and DDAH activity but also increase plasma ADMA concentrations in rats. Furthermore, atorvastatin treatment increased the expression of both SREBP1 and DDAH1 in thoracic aorta and decreased plasma ADMA levels in insulin-resistant rats. These results suggest that atorvastatin may protect endothelial function by modulating the DDAH1/ADMA system, specifically by restoring DDAH activity in insulin-resistant rats. Whether SREBP1 may act as a mediator in the regulation of DDAH1/ADMA by atorvastatin needs to be addressed in further studies.
Footnotes
Funding
This work was supported by: (1) Chinese Medical Association (CMA), grant number: 08010008; (2) PhD Programs Foundation of Ministry of Education of China, grant number: 200805331178; (3) Health Department, Hunan Province, China, grant number: B2011-010; (4) International Atherosclerosis Society, China Branch, grant number: NMF2008-003.
Conflict of interest statement
The authors state no conflict of interest.