
Abstract
Erythromelalgia is a rare clinical syndrome characterized by intermittent heat, redness, swelling and pain more commonly affecting the lower extremities. Symptoms are mostly aggravated by warmth and are eased by a cold temperature. In some cases, symptoms can be very severe and disabling. Erythromelalgia can be classified as either familial or sporadic, with the familial form inherited in an autosomal dominant manner. Recently, there has been a lot of progress in studying Na(v)1.7 sodium channels (expressed mostly in the sympathetic and nociceptive small-diameter sensory neurons of the dorsal root ganglion) and different mutations affecting the encoding SCN9A gene that leads to channelopathies responsible for some disorders, including primary erythromelalgia. We present a severe case of progressive primary erythromelalgia caused by a new de novo heterozygous missense mutation (c.2623C>G) of the SCN9A gene which substitutes glutamine 875 by glutamic acid (p.Q875E). To our knowledge, this mutation has not been previously reported in the literature. We also provided a short literature review about erythromelalgia and Na(v) sodium channelopathies.
Case report
A 15-year-old female presented with a 13-year history of chronic intermittent painful skin redness and swelling of both feet and lower legs.
Her problem began when she started to walk. She had difficulty with, and avoided walking on rough textures and warm surfaces. She also cried when rough textured clothing was placed on the lower legs and feet. At the age of 4, the patient started to have frequent episodes of pain in the lower legs and feet with associated erythema, edema and warmth. Symptoms were present more often during the day and were aggravated by warm temperatures. She found symptomatic relief by immersing her feet in cold water. There were no other reported skin abnormalities or complaints, including joint pains, or constitutional symptoms including weight loss, fever, chills or fatigue. Her family history was negative for similar symptoms or vascular disorders.
For the subsequent 9 years, the patient had continued to have similar symptoms intermittently that did not affect her daily activities, school performance, or function. She was later diagnosed with hypothyroidism that was easily controlled with thyroid hormone supplement. Her exam was otherwise unremarkable with the exception of her small stature, placed on the fourth and 37th percentiles respectively for height and weight. The patient was diagnosed with growth hormone deficiency. IGF-1 values were low at 218 ng/ml (range: 228–957). She underwent growth hormone stimulation testing with clonidine, which showed her peak growth hormone response was 0.5 ng/ml (normal peak is greater than 10). A sustained trial of growth hormone replacement was not done at the family’s preference.
At the age of 13 years, the patient started to have intermittent blood pressure elevations that were worse at the times of her symptoms (highest systolic around 180). She used to receive intravenous nitroprusside that seemed to alleviate her skin symptoms. Her secondary hypertension work-up revealed normal electrolyte panels, urinalysis, renal ultrasound, and cardiac echocardiograms. However, during painful crises, plasma free total metanephrine (350 pg/ml; ref. range: < 206) and plasma free normetanephrine (324 pg/ml; ref. range: < 149) were noted to be increased. As part of work-up for neuroblastoma, CTs and MRIs of the abdomen, pelvis, and chest, MAG3 renograms, and I-123 MIBG scans were sequentially performed over an 8-year time period and were normal. Our presumption is that her periodic hypertension and mildly elevated metanephrines/normetanephrines were related to pain, although sufficient pain control could not be obtained to perform baseline testing when symptom-free. She was continued on oral phenoxybenzamine that was discontinued a few months later upon spontaneous improvement of her blood pressure.
In the past year (as she started puberty), her symptoms escalated in frequency and severity, evolving into constant erythema and warmth of the lower extremities with associated pain (Figure 1). Over the last few months the skin erythema vacillated to a dark blue discoloration. Symptoms were aggravated more by exertion, stress, weight-bearing and gravity dependency. She spent most of her time immersing her feet in cold water or exposing them to a fan. She could not wear socks or shoes, or cover her feet or legs. Her symptoms started to affect her activities of daily living and her school performance and attendance.
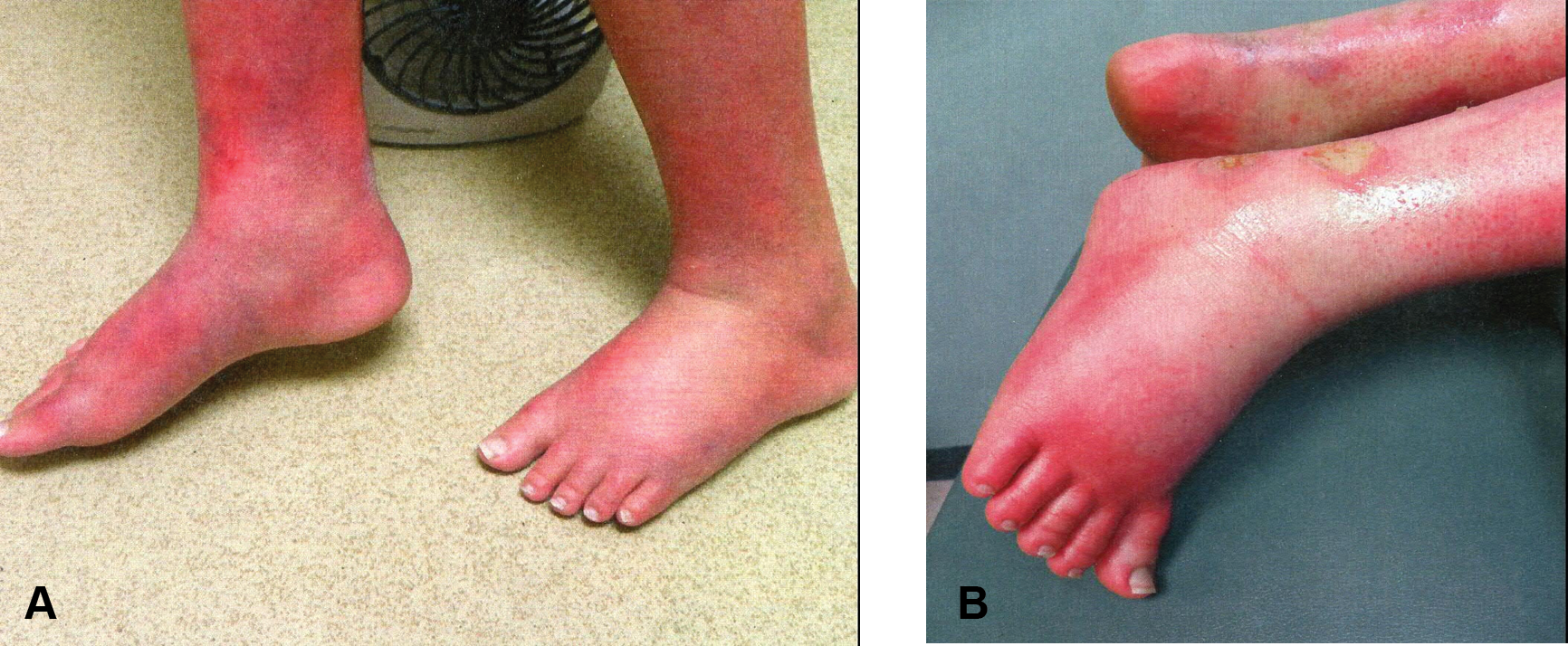
(A) / (B) Severe erythema and swelling of the lower extremities.
Laboratory evaluation of the patient was performed at the initiation of symptoms in early childhood, including enzyme testing for Fabry’s disease and autoimmune and inflammatory arthritides markers, and was found to be negative. Her most recent laboratory work revealed normal values: WBC 4.5 × 10 9 /l, Hgb 13.6 g/dl, PLT 269 × 10 9 /l, ESR 23 mm/h, CRP < 3 mg/dl, Na 141 mmol/l, K 4.7 mmol/l, creatinine 0.7 mg/dl, and TSH 3.6 mIU/l. There was slight elevation of Ca (10.5 mg/dl) and ALT (47 U/l). Lipid profile, iron studies and urinalysis were normal. Direct immunofluorescence of a skin biopsy was negative and H&E of the specimen showed a subcorneal non-specific inflammation.
The chest X-ray, ECG, ECHO and pulmonary function tests were normal. Autonomic reflex screen testing including heart rate responses and quantitative sudomotor axon reflex test (QSART) revealed evidence of generalized postganglionic sudomotor and mild adrenergic vasomotor dysfunction likely consistent with diffuse small fiber neuropathy. Thermoregulatory sweat testing revealed generalized anhydrosis. EMG showed no evidence of large fiber neuropathy. Baseline lower extremities physiologic arterial studies revealed normal Doppler signals, increased temperature of the toes (but not exceeding core temperature), increased laser signals from a few digits and normal transcutaneous oxygen pressure tracing (TCPO2). With symptoms, she had similar findings with the exception of significantly decreased TCPO2 on the left foot.
Genetic testing revealed a heterozygous missense mutation p.Q875E (c.2623C>G) of the SCN9A gene (NM_002977.2), which substitutes glutamine 875 by glutamic acid and which has not been previously described in patients with erythromelalgia (Figure 2). The patient’s healthy parents and sibling were tested for the mutation and found to be normal, suggesting a novel de novo mutation. Of note, the residue at position 875 of the protein is highly conserved throughout evolution.
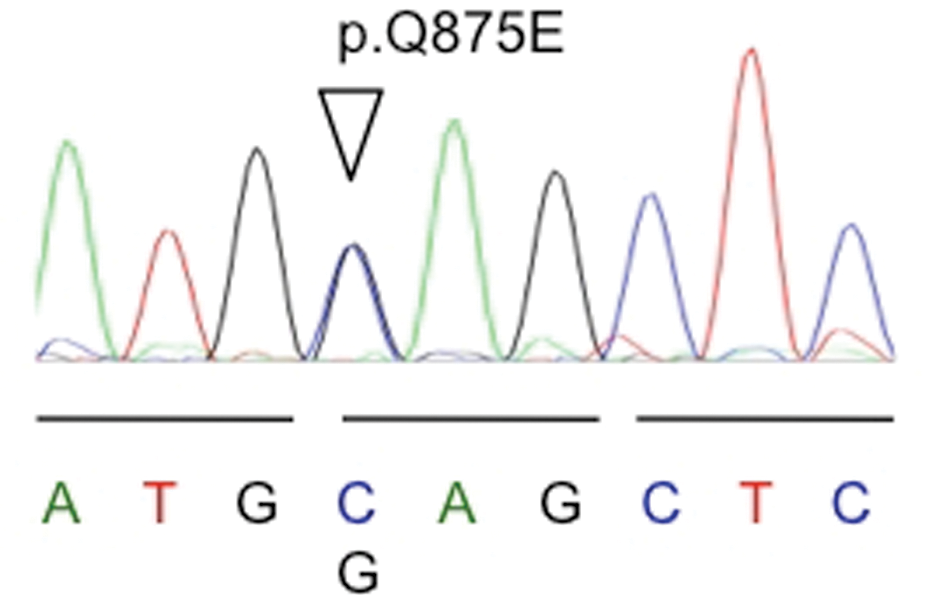
Sequence electropherogram of the heterozygous missense mutation p.Q875E (c.2623C>G) of the SCN9A gene.
Based on her clinical presentation and extensive work up, the patient was diagnosed with primary erythromelalgia. She had failed many therapies including aspirin, atenolol, systemic and topical lidocaine and steroids, clonazepam, narcotics, homeopathic therapies, acupuncture, TENS unit, and a re-trial of nitroprusside. Clonidine has helped some of the bedtime symptoms. She was started later on gabapentin, and topical amitriptyline-ketamine-Vanicream™ with some symptom relief.
Discussion
Erythromelalgia, or Mitchell’s disease, was named by and after Weir Mitchell in 1878 from erythro (red), melos (extremity), and algos (pain). 1 Graves had described the disease symptoms of hot and painful legs earlier, in 1834. 2 Other names used in the literature included acromelalgia from acro (tip end or extremity), erythralgia, erythermalgia from therme (heat), erythermomelalgia, and erythroprosopalgia from prosopon (face).3,4 Erythromelalgia is the most commonly used name to describe a rare clinical syndrome characterized by intermittent heat, redness, swelling and pain more commonly affecting the lower extremities that can be exacerbated by warming, exercise and dependence on legs, and relieved by cooling and elevation. 5
The pain is usually described as burning or piercing and sometimes can be very severe and disabling. Symptoms are usually intermittent but can be constant with changing intensity. It usually involves the lower extremities, occasionally affecting the hands and very rarely the ears and face. 6 Symptoms are more common in the summer and can be exacerbated by heat, ambulation, physical activity, sitting, leg dependence, or wearing shoes or gloves. 7 On the other hand, a cool temperature may alleviate symptoms and in some cases abort an episode. Symptoms tend to lessen with cooling of the extremities by immersion in ice or cold water; exposure to air current, such as by a fan; or elevation and uncovering the affected areas. 8
The incidence of the disease is very rare. A mean incidence of 1.3 per 100,000 was reported in persons living in Olmsted County, MN, USA in 2009. Women are slightly more affected than men. 9 Erythromelalgia can occur as a primary or secondary disorder. Primary erythromelalgia is mostly caused by mutation of the voltage-gated sodium channel α-subunit gene SCN9A. It can be classified as either familial or sporadic, with the familial form inherited in an autosomal dominant manner. Both of these may be further classified as either juvenile or adult onset. Juvenile onset occurs prior to age 20 years and frequently prior to age 10.10,11 Reported secondary causes of erythromelalgia may include some myeloproliferative diseases, blood dyscrasias, drugs, infections, malignancies, connective tissue and autoimmune diseases, cellular storage diseases, neuropathies, and some ingested materials (Table 1). 12
Reported causes of secondary erythromelalgia12

Although the exact pathophysiology remains unknown, it seems to be different in the two types. 13 At the molecular level, primary (inherited) erythromelalgia is an autosomal dominant disorder caused by gain-of-function mutations in the SCN9A gene encoding the Na(v)1.7 sodium channel expressed mostly in the sympathetic and nociceptive small-diameter sensory neurons of the dorsal root ganglion that leads to altered function. 14 The Na(v)1.7 sodium channel comprises 1988 amino acids (according to NM_002977.2) organized into four domains (D1–D4), each with six transmembrane segments (S1–S6). Segments S4 are probably the voltage-sensors and are characterized by a series of positively charged amino acids at every third position (Figure 3). 15 The amino acid 875 affected by our patient’s mutation is part of the fifth transmembrane span in D2. To date, around 20 mutations have been identified in patients with primary erythromelalgia (Table 2).16–27 Most of the mutations identified so far cluster within D1 and D2 of the protein. Some mutations are located in the cytoplasmic linkers, and others, like the one reported here, are located at the transmembrane domains of the channel.19,28 Functionally, most of the studied primary erythromelalgia-related mutations were shown to produce a hyperpolarizing shift in activation and slow deactivation and enhance the channel response to small depolarizing stimuli; changes that can confer hyperexcitability of cells harboring these channels. 15 While the alteration of the sympathetic neurons’ function leads to the microvascular symptoms, the alteration of the nociceptive neurons’ function results in severe burning pain, characterizing erythromelalgia. 10
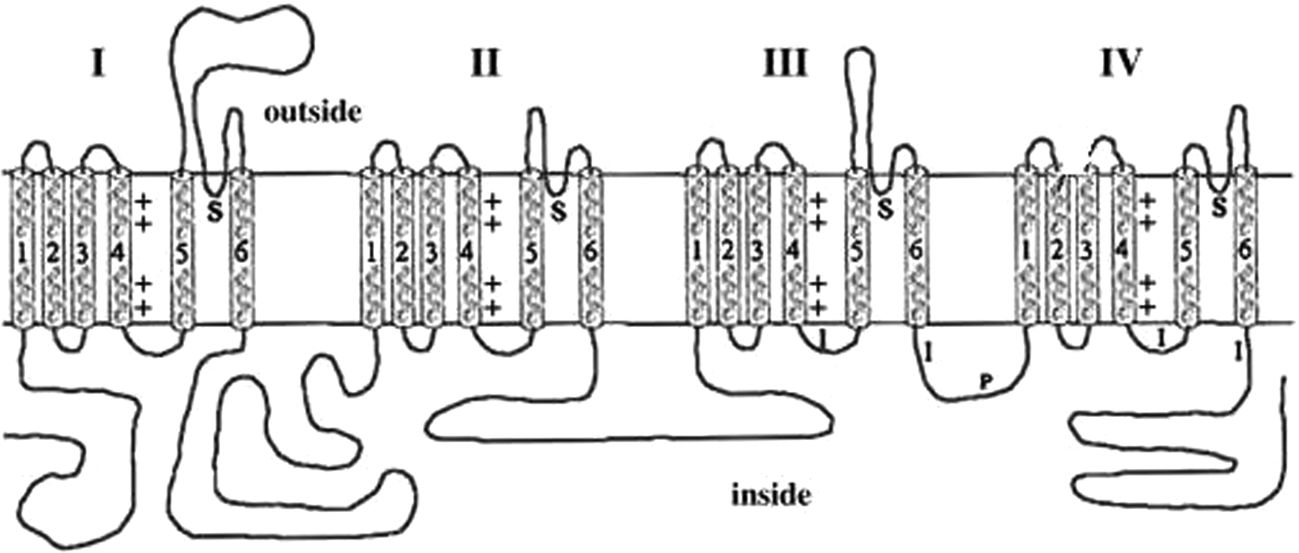
Na(v)1.7 sodium channel structure.
Sodium channel missense mutations at different nucleotide positions and resulting disorders
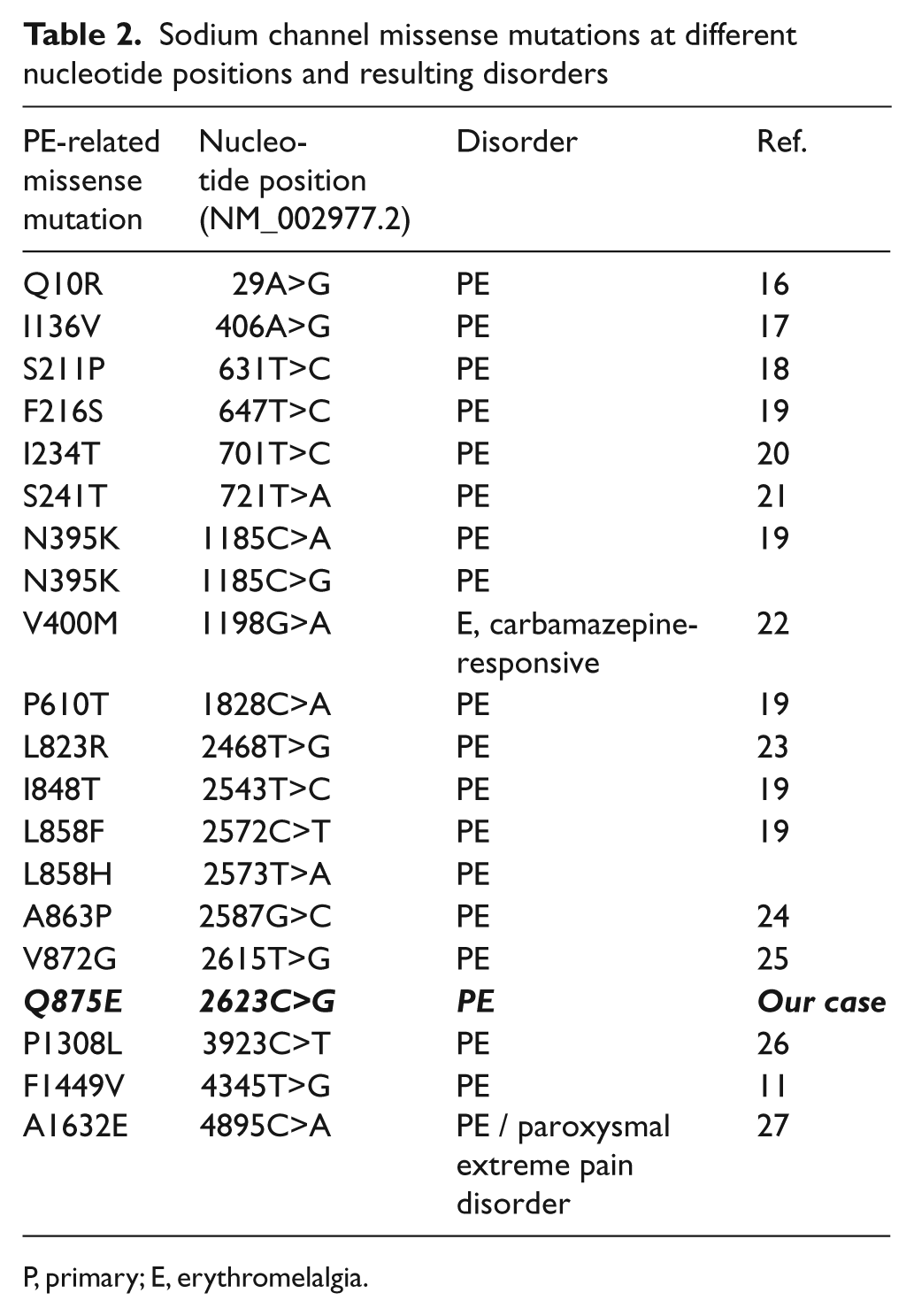
P, primary; E, erythromelalgia.
Despite an increase blood flow (as measured with laser Doppler), there is local hypoxia that can be reflected by low TCPO2 values, which might be explained by arteriovenous shunting at a microvascular level. 29 Unlike the primary type, secondary type pathophysiology is poorly understood and thought to be due to neuropathological and microvascular functional changes caused by the underlying condition. 30
Since there is no diagnostic test for erythromelalgia, a very good history and physical exam are crucial for the diagnosis. Differential diagnoses may include cellulitis, erysipelas, dermatitis, osteomyelitis, complex regional pain syndrome, systemic lupus erythematosus (SLE), peripheral neuropathy, arterial or venous insufficiency, and gout. 12 Vascular laboratory studies might reveal normal arterial Doppler signals, increased temperature and laser Doppler values in the presence of low TCPO2. 12 Autonomic reflex screen testing can be abnormal with small fiber disease. Genetic testing can be helpful in making the diagnosis of the primary type.10,11 There are no general guidelines on testing for SCN9A mutations. Testing can be considered in young patients with positive family history when secondary etiologies are less likely. Sometimes genetic testing is requested by the patient. Testing can also have some impact on family planning since the probability to inherit the mutation and having a child with the same disease is 50%. Genetic testing to prove inherited rather than secondary erythromelalgia might also limit repeated extensive and expensive work up looking for other underlying etiologies. So far, there is no specific therapy (e.g. Nav1.7 channel blockers) but once they are available, this will be a promising therapeutic approach for patients with positive mutations.
Management is very difficult and should follow a multidisciplinary approach. There is no single effective treatment for erythromelalgia. Management includes patient education, avoiding aggravating factors, cooling techniques, controlling secondary and underlying factors, and selective medications. 31 Aspirin is more effective in patients with secondary erythromelalgia caused by myeloproliferative disorders.12,31 Sodium nitroprusside may be helpful in children. 32 Vasoactive drugs including β-blockers, magnesium, prostaglandin E1, iloprost (prostacyclin analog), and ergot alkaloids have been reported to relieve symptoms.12,31 There are conflicting reports about the role of calcium channel blockers. 12 Neuroactive drugs including SSRIs, tricyclic antidepressants, gabapentin, pregabalin and benzodiazepines have also been reported to have some efficacy.12,31 NSAIDs and other types of analgesics and narcotics administered by different routes can be carefully used for pain control. 12 Surgical procedures including sympathectomy and sympathetic nerve block can be tried in intractable cases.12,33 There has been conflicting evidence for other therapies including acupuncture, biofeedback, hypnosis, and magnets.19,34
Our patient had severe progressive primary erythromelalgia caused by a new de novo heterozygous missense mutation (c.2623C>G) of the SCN9A gene which substitutes glutamine 875 by glutamic acid (p.Q875E). To our knowledge, this mutation has not been previously reported in the literature. Since the mutation occurred de novo and the residue at position 875 of the protein was highly conserved, this mutation is mostly the cause of her disease. Moreover, it was predicted to be damaging based on in-silico analysis using PolyPhen and Mutation Taster. We propose that p.Q875E, in line with the mutations described so far, is likely to cause a gain-of-function in the SCN9A gene. Brachydactyly is unreported in primary erythromelalgia and it may represent an unrelated finding. Short stature and growth hormone deficiency is not frequently reported in primary erythromelalgia. However, erythromelalgia, growth hormone deficiency, and hypertension responsive to recombinant human growth hormone have been reported by Cimaz et al. in 2001. It remains unknown whether or not this could represent a specific feature of this novel mutation. 35 Her disease exacerbation during puberty might implicate a hormonal role in the disease pathophysiology.
Pain-associated channelopathies represent an emerging field, and their discovery and understanding will pave the way for new management options.
Footnotes
This research received no specific grant from any funding agency in the public, commercial, or not-for-profit sectors.
The authors declare that there is no conflict of interest.