
Abstract
Background:
Siponimod, a sphingosine-1-phosphate (S1P) receptor modulator, reduces relapses and delays disability progression in patients with active progressive multiple sclerosis (MS).
Objective:
EXCHANGE assessed the safety/tolerability of siponimod in patients with advancing relapsing MS (RMS) converting from other disease-modifying therapies (DMTs).
Methods:
This 6-month, open-label, multicenter, single-arm, phase 3b study (NCT03623243) enrolled 185 patients with advancing RMS previously treated with other DMTs for ⩾3 months. Patients were converted to siponimod via a 6-day dose-titration regimen, or converted immediately, depending on prior DMT use.
Results:
Treatment-related adverse events (AEs) were reported by 31.9% (59/185) of patients, with headache (8.1%, n = 15), dizziness (3.8%, n = 7), and nausea (3.2%, n = 6) most commonly reported. Overall, an increase in heart rate (HR) 6 hours following the first dose of siponimod was observed (+2.47 bpm [0.66; 4.29]; p = 0.008). Patients switching from fingolimod without dose titration experienced no change in HR. Serious AEs were reported by 4.9% (9/185) of patients, and 8.6% (16/185) of patients discontinued the study treatment due to AEs.
Conclusion:
Conversion to siponimod from other DMTs was found to be generally well tolerated. Patients switching from other S1P-receptor modulators may be able to immediately transition to the siponimod maintenance dose without effects on HR.
Clinical Trial Registration:
ClinicalTrials.gov: NCT03623243 (https://clinicaltrials.gov/study/NCT03623243)
Keywords
Introduction
Multiple sclerosis (MS) is an immune-mediated central nervous system (CNS) disease characterized by inflammation, demyelination, and axonal/neuronal destruction that leads to severe disability. 1
Nine classes of disease-modifying therapies (DMTs) are currently indicated for use in MS. 2 Switching DMTs may be necessary due to disease activity, safety and tolerability, family planning, or payor access. In a study published in 2019, almost 50% of patients with MS switched DMTs within the previous 3 years. 3
Siponimod, an oral sphingosine-1-phosphate (S1P)1/S1P5-selective S1P receptor antagonist, was approved for use in MS by the US Food and Drug Administration in 2019 and the European Union in 2020.4,5
The phase 3 registration trial EXPAND showed siponimod significantly decreased disability progression in patients with secondary progressive MS versus placebo. 6 EXPAND showed that the safety profile of siponimod was aligned with other S1P modulators, including more frequent observations of bradycardia at treatment initiation with siponimod (4%) versus placebo (3%). To date, studies have not focused on safety considerations in patients who convert to siponimod from other approved DMTs.
Here, we describe the results of the EXCHANGE trial investigating the safety and tolerability of switching to siponimod from another approved DMT in patients with advancing relapsing MS (RMS).
Materials and methods
Study design
EXCHANGE (NCT03623243) was a 6-month, open-label, multicenter, single-arm study in patients with advancing RMS from 63 sites in the United States (Figure 1).7,8 The study consisted of a 28-day screening period, 6-month core treatment period, and telephone call 30 days after the end of treatment. Assessment visits were conducted at screening and again at days 1, 28, 84, and 168 of the treatment period. Patients could receive commercial supply of siponimod after the end of the study.
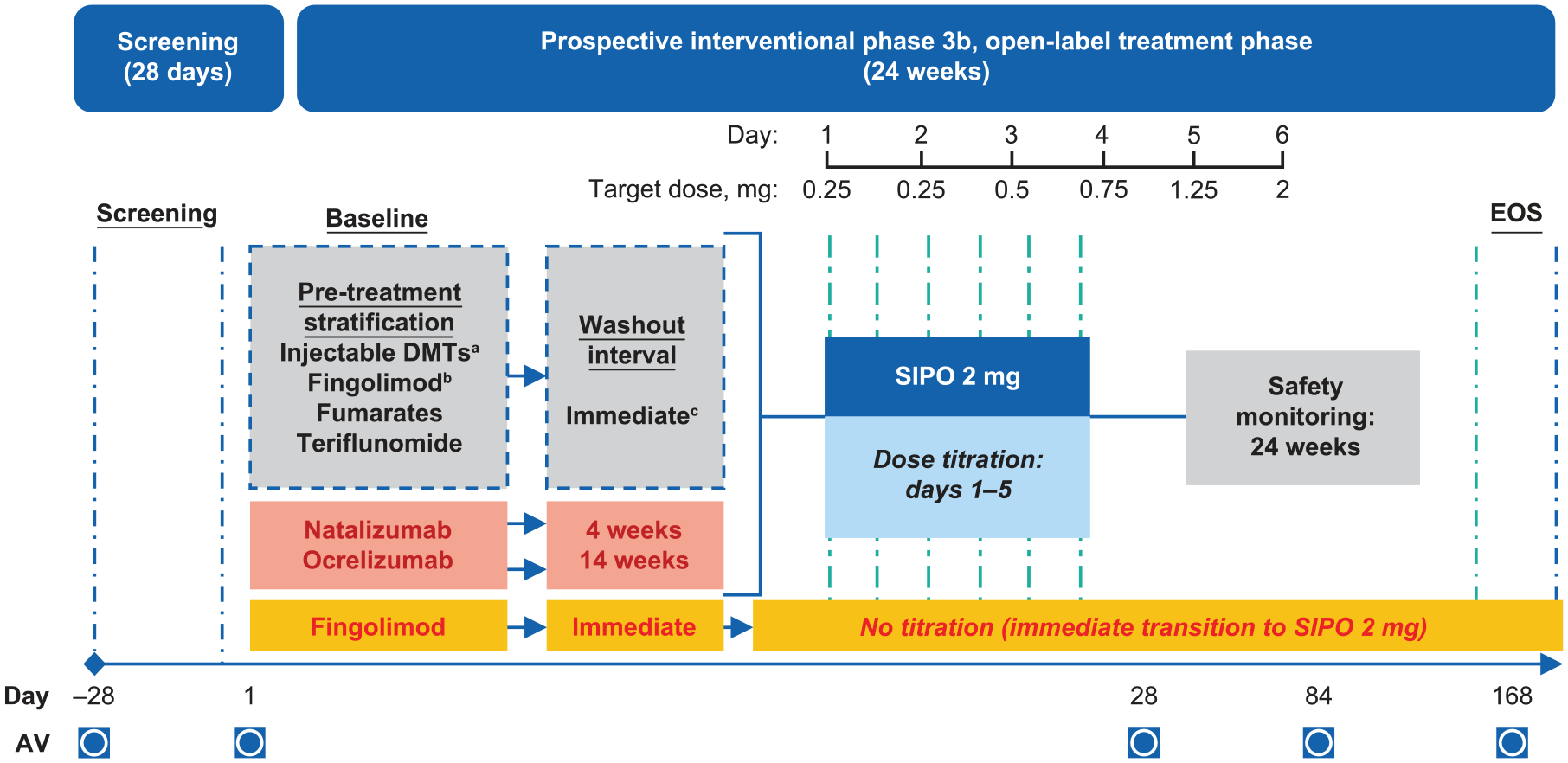
Study design of EXCHANGE.
The study was conducted according to the principles of International Council for Harmonization E6 Guideline for Good Clinical Practice and the Declaration of Helsinki. Informed written consent was obtained before any study procedure was performed.
Study population
The study population consisted of men and women, 18–65 years of age, with an Expanded Disability Status Scale (EDSS) score 2.0–6.5, and with advancing RMS. For the purposes of this study, we developed an operational definition of advancing RMS to help identify patients whose MS initially followed a relapsing-remitting disease course and who then, at some point, demonstrated onset of relatively fixed impairment. EDSS > 2 defines the beginning of relatively fixed impairment. This further impairment could be indicative of either worsening activity or disease progression. Worsening activity denotes an increase in neurological dysfunction and/or disability, with or without evidence of relapses or disease activity on magnetic resonance imaging (MRI). In addition, disease progression denotes the continuous or steady worsening of neurological impairment over ⩾6 months not explained by incomplete recovery from relapses. Inclusion of patients under the above operational definition of advancing RMS was at the discretion of the investigator based on perceived worsening over the period prior to screening.
Patients must have been previously treated with another approved MS therapy for ⩾3 months immediately prior to enrollment.
A comprehensive list of inclusionary and exclusionary criteria can be found in the Supplementary Material.
Treatments
Patients initiated siponimod within 24 hours of cessation of their previous DMT and used a 5-day dose escalation. Exceptions included: patients who previously received teriflunomide underwent accelerated elimination via administration of oral cholestyramine (8 g) three times per day for 11 days before starting siponimod, fingolimod-treated patients who joined the study ⩾11 months after recruitment initiation immediately converted to the target daily maintenance dose of siponimod (2 mg) without dose titration, and patients previously treated with natalizumab or ocrelizumab underwent at least a 4- or 14-week washout period, respectively, before starting siponimod. On day 1 (baseline), heart rate (HR) was monitored and measured using Holter telemetry after administration of the initial dose of siponimod. Full details of the washout regimens for each prior DMT can be found in Supplementary Table 1.
Except as noted above, a 5-day dosage escalation included the following: days 1 and 2, 0.25 mg; day 3, 0.5 mg; day 4, 0.75 mg; day 5, 1.25 mg; and day 6 and beyond, 2 mg.
Treatment compliance (defined as [duration of exposure − number of days marked as dosing error and/or the dose was not administered] *100/duration of exposure; duration of exposure = last known date of siponimod administration – first date of siponimod administration + 1) was assessed by the investigator and/or study site personnel at each visit using pill counts and information provided by the patient/caregiver. During the maintenance treatment period, patients with <80% compliance were counseled accordingly.
Study outcomes
The primary outcome was the occurrence of any treatment-related adverse events (TRAEs) during the 6-month siponimod treatment period. TRAEs were defined as any AE which the study investigator suspected to be related to study medication.
Secondary outcomes were treatment satisfaction, treatment persistence, and cardiac safety during siponimod initiation. Treatment satisfaction was determined by the change from baseline in the abbreviated 9-item Treatment Satisfaction Questionnaire for Medication (TSQM-9), a validated tool that includes the domains of Effectiveness, Convenience, and Global satisfaction. 9 Treatment persistence was measured by patient retention. Cardiac safety was evaluated via change from baseline in HR 6 hours after administration of the first dose of siponimod and overall occurrence of any AEs.
Exploratory objectives included number of relapses (as determined by the study investigator), the effect of siponimod on cognition, serum neurofilament light chain (sNfL) levels, and other measures of disease evolution over the course of the 6-month study. NfL was measured using the ADVIA Centaur® sNfL assay (Siemens Healthineers, Forchheim, Germany). Cognition was evaluated using the Processing Speed Test (PST), a validated, self-administered, iPad-based tool that measures processing speed similarly to the Symbol Digit Modalities Test. 10 Patient-reported functional status was measured using the Patient-Determined Disease Steps (PDDS) scale, a validated questionnaire measuring patient-reported disability on a scale from “normal” to “bedridden.” 11
Statistical analyses
All analyses were performed using all patients who received ⩾1 dose of siponimod.
Sample size calculations were based on the expected rate of TRAEs over 6 months. Because study subjects were expected to be more similar to the patients with relapsing-remitting MS (RRMS) studied in the fingolimod FREEDOMS (NCT00289978) and TRANSFORMS (NCT00340834) trials, pooled data from these trials were used to derive an expected AE rate of 45%. A planned sample size of 300–400 patients provided a margin of error ranging from 4.9% to 5.6% of the estimated rate of AEs. With the final sample size of 185 patients, a margin error of 7.2% is estimated.
Demographic and other baseline data were summarized descriptively.
For the primary endpoint, the number (and percentage) of patients with TRAEs were summarized by Medical Dictionary for Regulatory Activities preferred term. A patient with multiple TRAEs within a category (overall, primary system organ class, or preferred term) was counted once toward the total of that category.
For secondary endpoints, summary statistics were provided for change in HR from baseline to 6 hours after first dose of siponimod, and a post hoc paired t-test was performed. Overall, AEs were summarized as described for the primary endpoint.
Summary statistics were provided for changes in TSQM-9 from baseline. For treatment persistence, the number (and percentage) of patients who completed the study were provided, along with the number of patients who discontinued the study.
For exploratory endpoints, annualized relapse rates (ARRs) were estimated using a negative binomial model, with age group as an explanatory variable and the logarithm of duration (in years) as an offset variable. For the PDDS, the number and percentage of stable/improved patients and patients worsening were reported. A complete-case analysis was conducted to assess the impact of missing data values. Geometric mean values of sNfL were estimated from fitting a post hoc mixed-effects model for repeated measures (MMRM) using categorical visit and with prior MS therapies, age, and sex as factors. Changes in PST were obtained from fitting a post hoc MMRM using categorical visits and with prior MS therapies, age, and sex as factors. The MMRM method to handle missing values uses all available data and produces unbiased estimates under the missing at random assumption. Complete-case analyses were considered supportive only.
COVID-19 sub-study
This sub-study assessed immune responses to non-live messenger RNA (mRNA) COVID-19 vaccines in a sub-group of siponimod-treated patients who had received ⩾2 doses of an mRNA COVID-19 vaccine. Patients with a known previous COVID-19 diagnosis or contraindication to receiving an mRNA COVID-19 vaccine were excluded.
Patients could be vaccinated before or after switching to siponimod. A sub-study visit (SV1) was planned ⩾14 days after the second vaccine dose. Severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) spike (S1) immunoglobulin G (IgG) was used to assess vaccine response and SARS-CoV-2 nucleocapsid (NCP) IgG was assessed simultaneously to identify confounding COVID-19 infection. For those who tested positive for anti-S1 IgG at SV1, anti-S1 serum IgG concentration was summarized.
Results
Demographic and baseline clinical characteristics
From February 2019 to July 2022, 342 patients were screened for eligibility and 185 patients were enrolled (Figure 2). This study was stopped early due to slow enrollment likely caused by several factors, including the COVID-19 pandemic. Mean (standard deviation [SD]) patient age was 46.5 (10.35) years, and most patients were female (74.1%) and White (84.9%) (Table 1). Most patients (75.1%) had RRMS at the start of the study and, in the 12 months prior to screening, approximately half had experienced ⩾1 relapse. The most common prior DMTs were oral therapies including fingolimod (28.6%), dimethyl fumarate (22.7%), and teriflunomide (16.2%) followed by injection therapies including glatiramer acetate (14.6%) and interferon-beta (12.4%; Table 1).
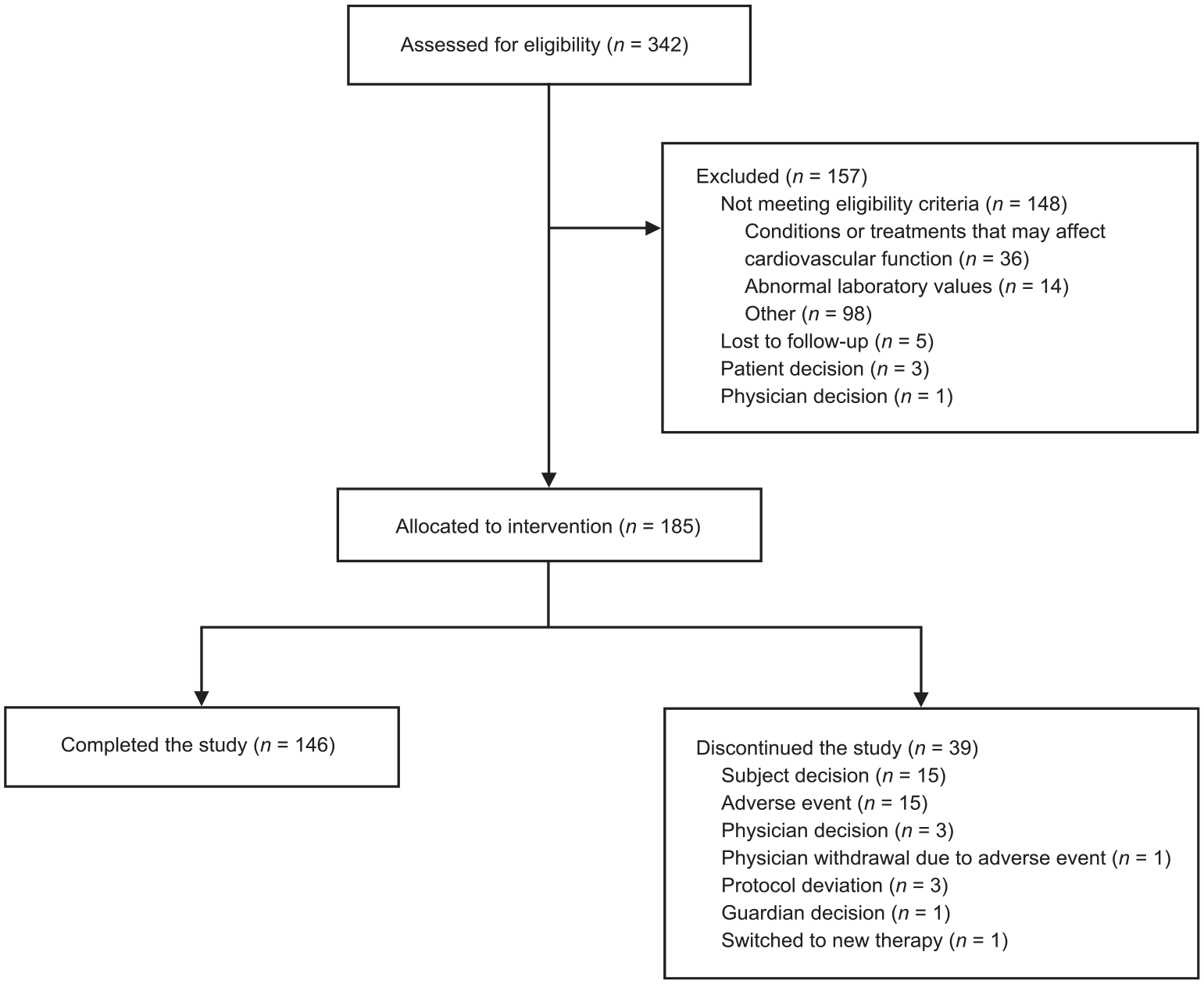
EXCHANGE patient flow chart.
Demographic and baseline clinical characteristics.
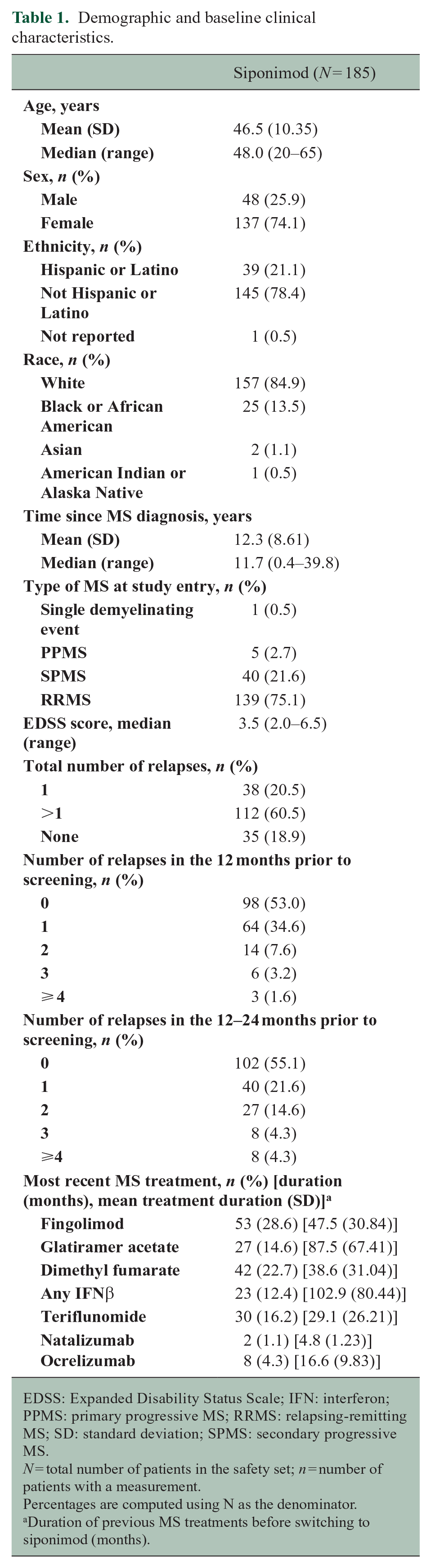
EDSS: Expanded Disability Status Scale; IFN: interferon; PPMS: primary progressive MS; RRMS: relapsing-remitting MS; SD: standard deviation; SPMS: secondary progressive MS.
N = total number of patients in the safety set; n = number of patients with a measurement.
Percentages are computed using N as the denominator.
Duration of previous MS treatments before switching to siponimod (months).
Median exposure to siponimod was 168 days (range: 1–198 days, interquartile range, 161-170), with 151 (81.6%) patients being exposed for ⩾20 weeks. In total, 146 (78.9%) patients completed the study and 39 (21.1%) patients discontinued (Figure 2).
Safety
AEs
During the study, 31.9% of patients experienced AEs considered by the investigator to be treatment related (Table 2). Suspected TRAEs occurring in ⩾3% of patients included headache (8.1%, n = 15), dizziness (3.8%), and nausea (3.2%) (Table 2). Higher frequencies of suspected TRAEs were observed when previous treatment was teriflunomide (15/30, 50%) or ocrelizumab (6/8, 75%) (Table 2). AEs occurring in ⩾5% of patients previously treated with teriflunomide include headache (4/30, 13.3%), lymphopenia (2/30, 6.7%), fatigue (2/30, 6.7%), and pain in an extremity (2/30, 6.7%). AEs occurring in ⩾5% of patients previously treated with ocrelizumab include headache (3/8, 37.5%).
AEs suspected to be related to the study medication in the safety set.

AE: adverse event; ALT, alanine aminotransferase; AST, aspartate aminotransferase; DMT: disease-modifying therapy; HR, heart rate; IFNβ: interferon-beta.
N = total number of patients in the safety set; n = number of patients with a measurement.
A patient with multiple AEs is counted only once in the “⩾1 AE” row.
Percentages are computed using N as the denominator.
N = total number of patients previously treated with DMT listed.
Including events that were not suspected by the investigator to be treatment related, 74.6% of patients experienced ⩾1 AE (Table 3). The most common AEs occurring in ⩾ 7% of patients included headache (13.5%), urinary tract infection (8.6%), nausea (8.1%), dizziness (7.0%), and fatigue (7.0%).
AEs in the safety set.

AE: adverse event; SAE: serious adverse event.
N = total number of patients in the safety set; n = number of patients with a measurement.
A patient with multiple AEs or SAEs was only once in the “⩾1 AE/SAE” rows.
Percentages are computed using N as the denominator.
AEs reported by ⩾5 patients.
Nine (4.9%) patients reported ⩾1 serious AE (SAE), including non-cardiac chest pain, appendicitis, cellulitis, aspiration pneumonia, pyelonephritis, respiratory fume inhalation disorder, cerebrovascular accident, hemiparesis, MS progression, MS relapse, tubulointerstitial nephritis, and lymphoedema (Table 3). Three SAEs in 2 patients were reported as being treatment related (ischemic stroke, pyelonephritis, and tubulointerstitial nephritis). The patient who experienced the ischemic stroke discontinued siponimod and recovered.
Most AEs were reported by patients within the first 4 weeks of the study (52.4%), with 39.2% of patients reporting AEs in weeks 4–12 and 40.1% in weeks 12–24.
Cardiac safety
In the overall population (n = 175), an increase in HR from baseline (73.12 [10.74] bpm) to 6 hours (75.59 [13.87] bpm) after the first dose of siponimod was observed (+2.47 bpm (0.66; 4.29); p = 0.008; Figure 3). Five patients (2.7%) experienced bradycardia during the titration period (Table 2). No specific pattern was observed when analyzed by prior DMT (Figure 3). In the small fingolimod sub-group who were switched to siponimod without dose titration, and for whom 6-hour HR data were available (n = 10), mean (SD) HR at baseline (74.20 [10.12] bpm) was unchanged 6 hours later (74.70 [13.99] bpm) (Figure 3).
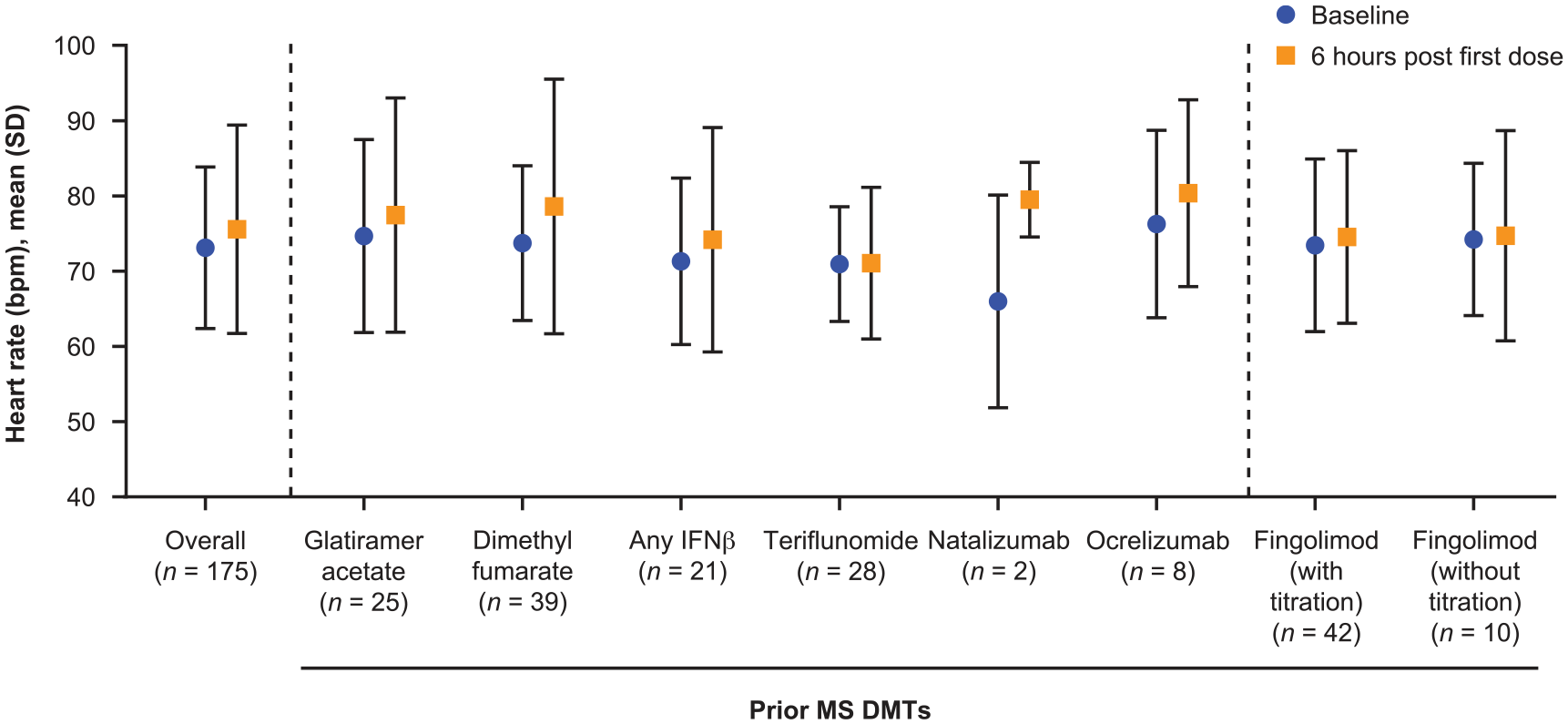
Mean HR at baseline and 6 hours post first dose by prior MS DMTs.
Hospitalizations
Nine (4.9%) patients reported ⩾1 hospitalization, of which 2 patients (1.1%) reported >1 hospitalization during the treatment period, including for appendicitis, aspiration pneumonia, cellulitis, hemiparesis, lymphoedema, non-cardiac chest pain, respiratory fume inhalation disorder, MS relapse, pyelonephritis, tubulointerstitial nephritis, MS progression, and cerebrovascular accident.
Efficacy
Treatment satisfaction
Mean TSQM-9 scores increased numerically at all subsequent visits versus baseline across all domains (Figure 4). Mean (SD) Convenience score increased from 69.6 (20.75) at baseline to 84.2 (15.22) at day 28 and 85.9 (14.37) at day 168. Mean (SD) Effectiveness score increased from 57.2 (20.67) at baseline to 67.3 (19.88) at day 28 and 71.0 (21.31) at day 168. Mean (SD) Global satisfaction score increased from 56.7 (21.98) at baseline to 67.2 (21.11) at day 28 and 72.4 (23.84) at day 168.
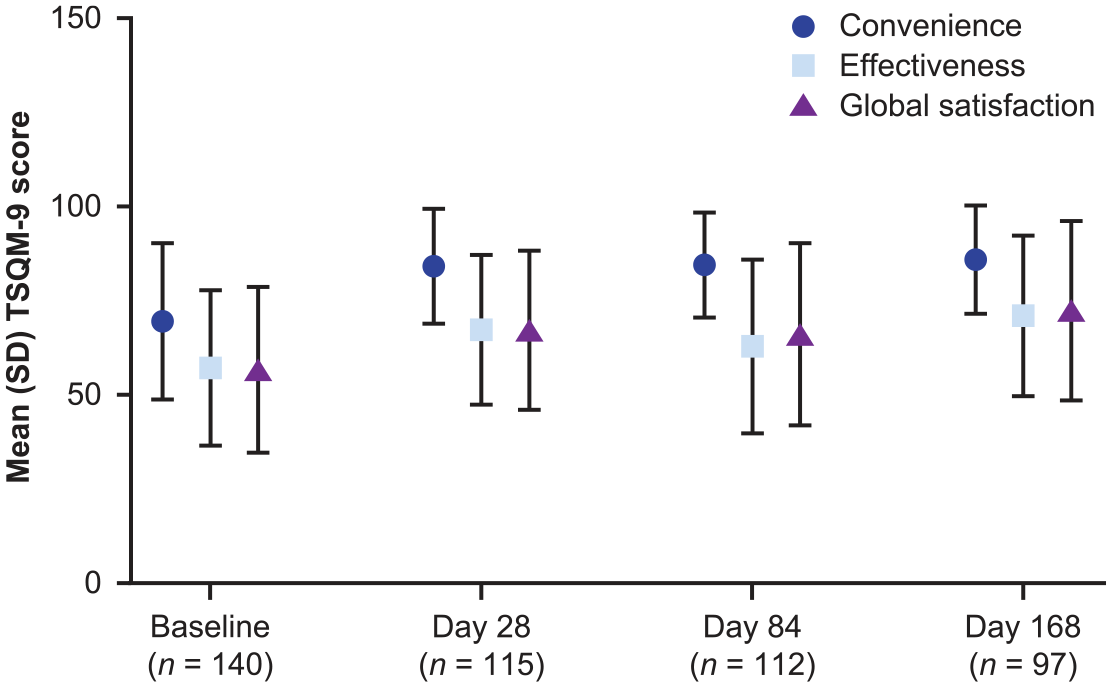
TSQM-9 scores for the domains of Convenience, Effectiveness, and Global satisfaction at baseline and days 28, 84, and 168. Analysis was based on observed data. No imputation was performed for missing data. n: number of patients with measurement; SD: standard deviation; TSQM-9: abbreviated 9-item Treatment Satisfaction Questionnaire for Medication.
Relapses
The estimated (95% CI) ARR was 0.17 (0.08–0.35). Out of 185 enrolled patients, 13 patients experienced a total of 14 confirmed relapses, that is, >90% of patients remained free of clinical relapses.
Cognition
Adjusted mean (95% CI) PST scores on days 1, 84, and 168 were 40.4 (37.8–43.0), 43.3 (40.7–46.0), and 46.7 (44.4–49.0), respectively (Figure 5(a)). Average PST score improved at days 84 and 168, with an adjusted mean increase of 2.9 (0.5, 5.3; p = 0.0181) at day 84 versus baseline and an adjusted mean increase of 6.3 (4.1, 8.5; p ⩽ 0.0001) on day 168 versus baseline (Figure 5(b)).
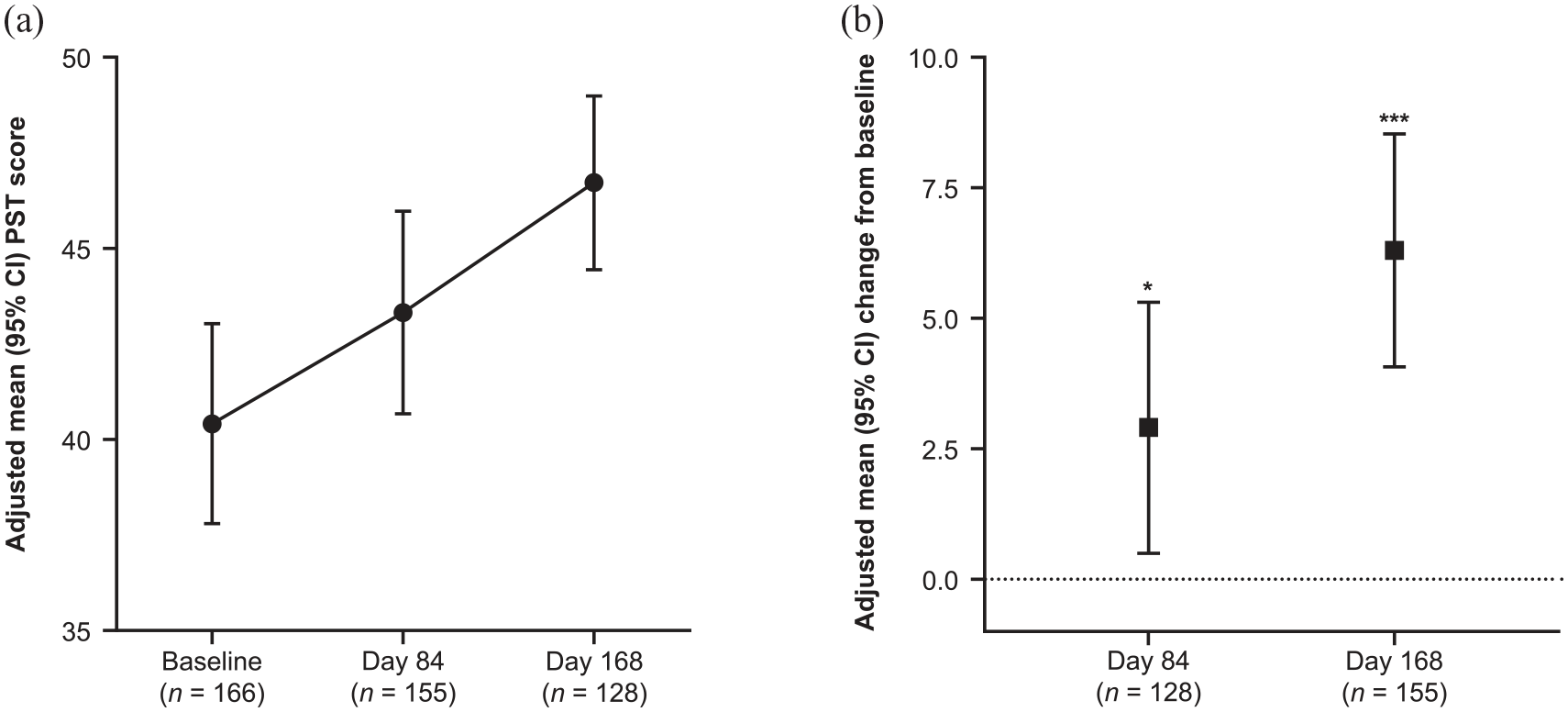
(a) PST scores at baseline, day 84, and day 168. (b) Change from baseline for PST score at day 84 and day 168. *p ⩽ 0.05, ***p ⩽ 0.0001. CI: confidence interval; n: number of patients with measurement; PST: Processing Speed Test.
NfL concentrations
Adjusted geometric mean (95% CI) NfL concentrations on days 1, 84, and 168 were 9.3 (8.6–10.1) pg/mL, 10.7 (9.9–11.5) pg/mL, and 8.9 (8.4–9.5) pg/mL, respectively (Figure 6(a)). The mean ratio for NfL concentration on day 84 versus baseline was 1.15 (1.08–1.21; p < 0.0001) and on day 168 versus baseline was 0.96 (0.90–1.02; p = ns) (Figure 6(b)).
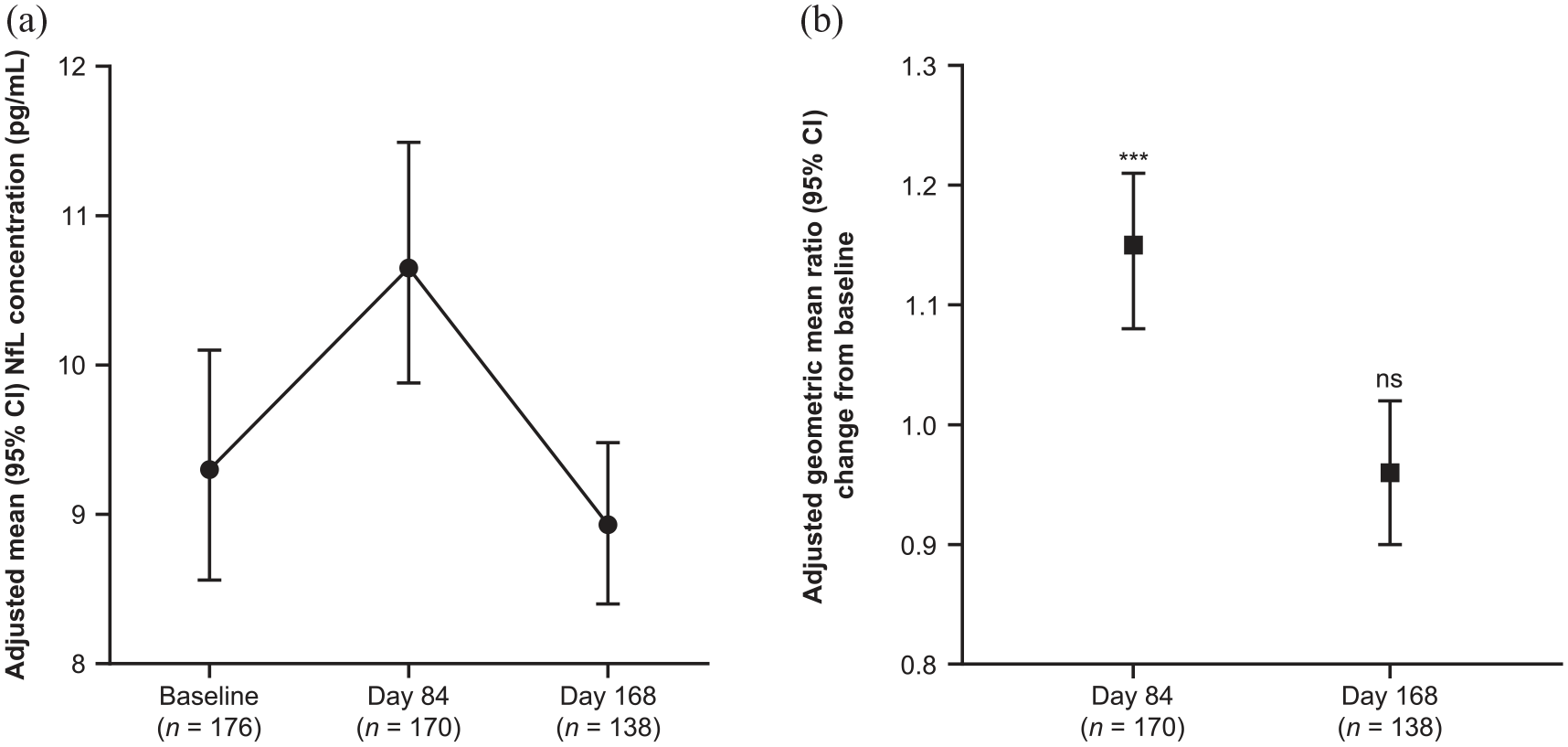
(a) NfL concentration (pg/mL) at baseline, day 84, and day 168. (b) Change from baseline in NfL concentration (ng/L) at day 84 and day 168. ***p ⩽ 0.001; ns = not significant.
Association between baseline NfL concentrations and relapses
The adjusted geometric mean (95% CI) baseline NfL concentration was 13.7 (8.4–22.5) in patients who experienced relapses (confirmed or unconfirmed) during the study and 9.0 (8.3–9.7) in patients who did not experience relapses during study.
Patient-reported disability
The majority of patients (73.5%) reported stable (49%) or improved (24.5%) PDDS over the study period, and 26.5% reported PDDS worsening. A complete-case analysis for PDDS (n = 89) showed similar results (Supplementary Figure 1).
COVID-19 vaccine sub-study
The patient demographics for the COVID-19 vaccine sub-study were similar to the core study (Table 4). Of the 11 sub-study patients, 4 (36.4%) were being treated with siponimod at the time of vaccination (Table 4). At SV1, 8 of 9 (88.9%) patients tested negative for anti-NCP serum IgG, indicating no prior exposure to the natural infection, and 7 (77.8%) patients tested positive for anti-S1 serum IgG, indicating that a majority of patients demonstrated a vaccine response with no prior natural exposure to COVID-19 (Table 4). In the 7 (77.8%) patients who tested positive for anti-S1 serum IgG, the mean (SD) titer was 1919.0 (1351.29) (Table 4).
COVID-19 vaccine sub-study.

DMT: disease-modifying therapy; FIN: fingolimod; GA: glatiramer acetate; IFNβ: interferon-beta; IgG: immunoglobulin G; NCP: SARS-CoV-2 nucleocapsid; OCR: ocrelizumab; SD: standard deviation; SIPO: siponimod; SV1: SARS-CoV-2 spike; TER: teriflunomide.
N = total number of patients in the COVID-19 vaccine sub-study; n = number of patients with a measurement; M = number of patients with anti-NCP and anti-S1 measurement.
Percentages are computed using N as the denominator unless otherwise stated.
Discussion
Patients with MS may switch treatments for a variety of reasons, including new disease activity and/or progression and safety or tolerability considerations. EXCHANGE was the first study to assess the safety and tolerability of converting patients from an approved oral, injectable, or infusion DMT to siponimod. The results of this study indicate that conversion to siponimod over a 6-day titration period was generally well tolerated, with no unexpected new safety findings. Although approximately one-third of patients reported TRAEs, most were mild in severity. SAEs were reported in nine patients, and no deaths occurred. Siponimod’s safety profile aligned with that observed in the phase 2 BOLD and phase 3 EXPAND studies,6,12 and other S1P receptor modulators including fingolimod,13,14 ozanimod,15,16 and ponesimod. 17 The 78.9% overall persistence with treatment was similar to the 82% observed in EXPAND. 6
Previous studies have shown cardiac first-dose effects with S1P therapies, including siponimod.6,12 These effects may be due to S1P receptor expression in cardiac myocytes 18 and are reduced using a dose-escalation regimen when starting siponimod. In EXCHANGE, there was no evidence of a reduction in mean HR when initiating siponimod in the overall group or in subgroups by prior DMT. The dose-titration regimen employed here could therefore mitigate the adverse cardiac events previously seen with siponimod initiation without dosage escalation, regardless of prior DMT usage. Particularly, transition to siponimod from fingolimod without dosage escalation was safe and well tolerated, suggesting that conversion to siponimod from other S1P receptor modulators may not require a dose-escalation regimen before initiating the full maintenance dose.
After switching to siponimod, numerical improvements were observed in treatment satisfaction across all TSQM domains; these improvements were generally maintained for the duration of the study. Although a clinically meaningful change in TSQM score has not been published, higher TSQM Convenience scores have been linked to greater treatment adherence rates in patients with MS. 19
By day 168, there was an improvement in cognitive functioning as measured by the PST. However, it is difficult to interpret these results due to the significant practice effects observed with the PST 10 and a lack of a verified indicator of a clinically meaningful change in score for this test. Although most patients also reported stable or improved disability as measured by PDDS, 26.5% reported worsening of PDDS.
An initial rise in sNfL concentrations was observed at day 84, with sNfL returning to baseline levels by day 168. sNfL levels after 12 and 24 weeks of siponimod treatment were comparable to treatment with other DMTs including interferon, teriflunomide, and anti-CD20s.20,21 The reason for the observed transient increase in sNfL is unclear but may be related to a transient return of MS disease activity during the washout period required for some previous DMTs and is therefore unlikely to be of clinical significance.
Study limitations
Although a sample size of up to 400 patients was originally targeted, the study ended prematurely with 185 patients. This increased the margin of uncertainty regarding AEs from ~5.6% to 7.2%.
As AEs were determined to be treatment related by each site investigator and were not independently verified, there is risk of bias and site-to-site variability in attribution. In addition, there was no blinding of each investigator as to treatment and no comparator employed.
The number of patients converting from natalizumab (n = 2) and ocrelizumab (n = 8) were small; thus, generalizing the experience of changing to siponimod from these therapies is limited. In addition, the short duration of EXCHANGE does not allow for assessment of AEs that may emerge with more prolonged observation. EXCHANGE was not designed to assess disease activity after siponimod discontinuation.
As with other single-arm studies, there are inherent limitations to the study design used in EXCHANGE, including the open-label design and potential selection bias that may favor patients expected to tolerate siponimod. These aspects may have contributed to the comparatively lower TRAE rate observed here compared with previous siponimod trials.
Conclusion
EXCHANGE demonstrated that conversion to siponimod from other approved DMTs was generally well tolerated and safe when utilizing a gradual dose-escalation regimen. The results also provide preliminary evidence that patients switching from other S1P-receptor modulators may be able to immediately transition to full-dose siponimod without adverse effects on HR; however, further accumulated clinical experience will supplement the results presented here.
Supplemental Material
sj-docx-1-msj-10.1177_13524585251330085 – Supplemental material for Safety and tolerability of conversion to siponimod from other disease-modifying therapies in patients with advancing forms of relapsing MS: Results from the EXCHANGE study
Supplemental material, sj-docx-1-msj-10.1177_13524585251330085 for Safety and tolerability of conversion to siponimod from other disease-modifying therapies in patients with advancing forms of relapsing MS: Results from the EXCHANGE study by Robert J Fox, Stanley Cohan, Yang Mao-Draayer, Bianca Weinstock-Guttman, Linda-Ali Cruz, Sophie Arnould, Gina Mavrikis Cox and Amit Bar-Or in Multiple Sclerosis Journal
Footnotes
Acknowledgements
Medical writing support was provided by Frankie Sorrell, PhD, of Envision Pharma Inc. and funded by Novartis Pharmaceuticals Corporation.
Data availability
Declaration of conflicting interests
The author(s) declared the following potential conflicts of interest with respect to the research, authorship, and/or publication of this article: R.J.F. has received personal consulting fees from AB Science, Biogen, Bristol Myers Squibb, Eli Lilly and Company, EMD Serono, Genentech, Genzyme, Greenwich Biosciences, Immunic, iNmune Bio, Janssen, Novartis, Sanofi, Siemens, and TG Therapeutics; has served on advisory committees for AB Science, Biogen, Immunic, Janssen, Novartis, and Sanofi; and has received clinical trial contract and research grant funding from Biogen, Novartis, and Sanofi. S.C. has received speaker fees from Biogen, Bristol Myers Squibb, Novartis, Roche/Genentech, and Sanofi-Genzyme; has served on advisory boards or as a consultant for Biogen, Bristol Myers Squibb, EMD Serono, Novartis, and Sanofi-Genzyme; and has received institutional (the Providence Brain and Spine Institute) research support from AbbVie, Adamas, Biogen, EMD Serono, MedDay, Novartis, Roche/Genentech, Sage Bionetworks, and Sanofi-Genzyme. Y.M-.D. has served as a consultant for and/or received grant support from: Acorda, Bayer, Biogen, Celgene/Bristol Myers Squibb, EMD Serono, Horizon, Janssen, Novartis, Questor, Roche/Genentech, Sanofi-Genzyme, TG Therapeutics, and Teva and has received grants from Chugai, the NIH NIAID Autoimmune Center of Excellence (UM1-AI110557-05, UM1 AI144298-01), Novartis, PCORI, and Sanofi-Genzyme. B.W-.G. has received consulting fees from Biogen, Bristol Myers Squibb, EMD Serono, Immunex, and SANA and has received research support from Biogen, Bristol Myers Squibb, EMD Serono, Genentech, and Novartis. L-.A.C. and G.M.C. are employees and stockholders of Novartis. S.A. is an employee and stockholder of Novartis AG. A.B-.O. has received personal fees for advisory board participation and/or consulting from Abata, Accure, Atara, Biogen, Bristol Myers Squibb/Celgene/Receptos, GlaxoSmithKline, Gossamer, Horizon, Immunic, Janssen/Actelion, Medimmune, Merck/EMD Serono, Novartis, Roche/Genentech, Sangamo, Sanofi-Genzyme, and Viracta; and has received grant support to the University of Pennsylvania from Biogen, Merck/EMD Serono, Novartis, and Roche/Genentech.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This study was funded by Novartis Pharmaceuticals Corporation. Novartis Pharmaceuticals Corporation supported the development of this manuscript and provided data analyses according to the direction of the authors.
Supplemental material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.