
Abstract
Background:
In multiple sclerosis, impact of treatment on disability progression can be confounded if treatment also reduces relapses.
Objective:
To distinguish siponimod’s direct effects on disability progression from those on relapses in the EXPAND phase 3 trial.
Methods:
Three estimands, one based on principal stratum and two on hypothetical scenarios (no relapses, or equal relapses in both treatment arms), were defined to determine the extent to which siponimod’s effects on 3- and 6-month confirmed disability progression were independent of on-study relapses.
Results:
Principal stratum analysis estimated that siponimod reduced the risk of 3- and 6-month confirmed disability progression by 14%–20% and 29%–33%, respectively, compared with placebo in non-relapsing patients. In the hypothetical scenarios, risk reductions independent of relapses were 14%–18% and 23% for 3- and 6-month confirmed disability progression, respectively.
Conclusion:
By controlling the confounding impact of on-study relapses on confirmed disability progression, these statistical approaches provide a methodological framework to assess treatment effects on disability progression in relapsing and non-relapsing patients. The analyses support that siponimod may be useful for treating secondary progressive multiple sclerosis in patients with or without relapses.
Keywords
Introduction
The defining characteristic of secondary progressive multiple sclerosis (SPMS) is disability progression independent of relapses. 1 However, relapses continue to occur, transition between multiple sclerosis phases is challenging to define and relapsing and progressive components may coexist for years. 2
In clinical trials, demonstrating treatment benefit on confirmed disability progression has proven elusive in SPMS populations.3–8 Following the EXPAND trial (NCT016665144), siponimod was approved in the United States for treatment of adults with relapsing forms of multiple sclerosis, including active SPMS.9,10 Siponimod significantly reduced the risk of disability progression and annualized relapse rate compared with placebo. 10
The question emerges as to what extent siponimod’s effect in reducing cumulative disability is independent of its impact on relapses. This is complicated by the occurrence of on-study relapses, the partial dependency of disability progression on relapses and the relapse-reducing effects of treatment.
Subgroup analyses based on pre-study or on-study relapses have been used; however, these approaches have shortcomings. The absence of relapse activity before enrolment does not predict on-study relapse activity, and the relapse-reducing effect of treatment introduces selection bias among non-relapsing individuals on study. Given that relapses are on-study events that may confound the effect of treatment on disability progression, we sought to disentangle treatment effects on progression from treatment effects on reducing relapse-associated disability using a novel statistical framework specifically designed for this purpose. 11
Patients and methods
EXPAND study design
EXPAND was a phase 3, randomized, double-blind, placebo-controlled trial, with an event-driven design and variable treatment duration of up to 3 years (median = 18 months). 10 EXPAND adhered to the International Conference of Harmonisation (ICH) for Good Clinical Practice and the Declaration of Helsinki. The protocol was approved by institutional review boards or ethics committees and patients gave written informed consent before the study. EXPAND investigated the effect of siponimod compared with placebo in a population consisting mostly of non-relapsing patients with SPMS.1,12 Overall, 64% of patients had not relapsed in the 2 years before enrolment and 87% did not relapse on study. Only 13% (60 of 462) of confirmed disability worsening events occurred in the setting of clinical relapses. The primary endpoint was reduction in 3-month confirmed disability progression (3mCDP) (see Kappos et al. 10 for eligibility and disability criteria).
Traditional subgroup analyses
Pre-specified subgroup analyses were undertaken based on the occurrence or absence of relapses in periods before or on study. Analyses for the subgroups with or without relapses in the 2 years before enrolment were previously reported. 10 A limitation of analyses based on post-randomization criteria is that they are subject to strong confounding factors, particularly when the criterion (presence of relapses) is affected by the drug under evaluation.
Statistical analyses
Analysis of estimands
Using draft guidance from the ICH, 11 we defined and estimated three estimands (Table 1): (1) treatment effect of siponimod in the subgroup (principal stratum (PS)) of non-relapsing patients (individuals who would not relapse regardless of treatment assignment); 13 (2) treatment effect of siponimod in a hypothetical scenario where relapses would not occur; and (3) treatment effect in a hypothetical scenario where siponimod had no effect on relapses. These estimands handle on-study relapses differently, and each contributes to an understanding of the effect of siponimod on disability progression independent of relapses.
Summary of post hoc analyses and their limitations.

IPCW: inverse probability censoring weight; PS: principal stratum.
PS estimand: disability progression in the subgroup of non-relapsing patients
The PS estimand was defined as (1) population: patients in EXPAND who would not relapse over a specified period irrespective of treatment assignment (‘non-relapsing stratum’); (2) variable: 3mCDP or 6-month confirmed disability progression (6mCDP); (3) intercurrent event: confirmed on-study relapse (captured by the population definition) during the period specified; and (4) population-level summary: risk ratio. 13
The non-relapsing stratum is one of four mutually exclusive PSs, each defined by the potential occurrence of a post-randomization relapse in a given period: (1) ‘non-relapsing’: PS of patients who would not relapse irrespective of treatment assignment; (2) ‘definite-relapsing’: PS of patients who would relapse irrespective of treatment assignment; (3) ‘benefitting’: PS of patients who would relapse if assigned to placebo but not if assigned to siponimod; and (4) ‘harmed’: PS of patients who would not relapse if assigned to placebo but would relapse if assigned to siponimod (Figure 1).
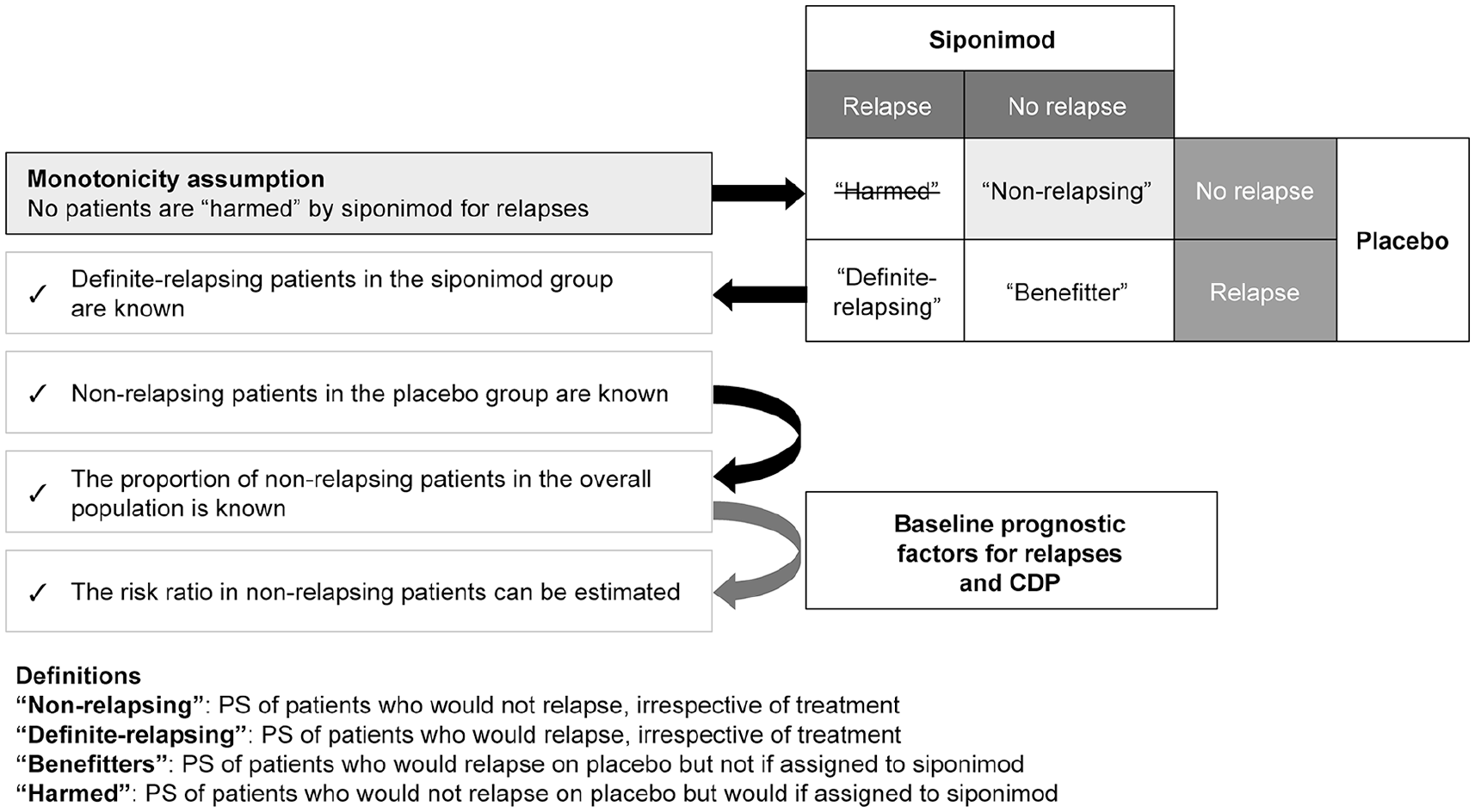
PS analysis: Estimation of the proportion of patients in each stratum using Bayesian logistic regression.
Membership within a PS is an inherent characteristic of a patient not confounded by treatment assignment, and can be considered similar to a pretreatment covariate. Thus, analysis of treatment effect in the non-relapsing PS yields a causal estimate not confounded by on-study relapses. 14 However, membership within a PS must be inferred; it is not possible to identify subjects of a PS directly because patients were not observed under both treatments.
Estimation of the probability of confirmed disability progression in the ‘non-relapsing’ PS was conducted separately by treatment group. For patients on placebo, the probability of confirmed disability progression was estimated based on individuals with confirmed disability progression who did not relapse on study. However, patients on siponimod who did not relapse on study could belong either to the ‘benefitting’ PS or to the ‘non-relapsing’ PS (Figure 1). Therefore, the contribution made by these patients to the overall estimated probability of confirmed disability progression was weighted by the probability that each patient belonged to the ‘non-relapsing’ PS.
Estimation used Bayesian logistic regression, adjusting for covariates considered prognostic for relapses and for confirmed disability progression (Expanded Disability Status Scale (EDSS) score at randomization, dichotomized as high (⩾6.0) or low (<6.0) and occurrence of relapses (yes/no) in the 2 years before randomization). Proportions of patients belonging to each PS and probabilities of confirmed disability progression were estimated within this covariate combination and combined with a weighted mean to obtain the overall probability of confirmed disability progression in the ‘non-relapsing’ PS. Prior distributions for all model parameters were described previously. 13
Following this approach, estimates for between-treatment risk ratios for confirmed disability progression were obtained for three different exposure periods, from randomization until (1) 12 months (minimum treatment duration post-randomization), (2) 18 months (median duration of treatment exposure), and (3) 24 months (longest duration for which interpretable data were available). Estimates are reported as posterior medians with 95% credible intervals. Additional analyses were conducted using 6mCDP as the disability endpoint and allowing for both unconfirmed and confirmed relapses.In addition, estimates for between-group risk ratios for confirmed disability progression were calculated at 12 and 24 months in two modified principal strata. In these strata, the definition of ‘non-relapsing’ was extended to include patients with no new or enlarging T2 lesions or those with fewer than two T2 lesions in magnetic resonance imaging (MRI) scans (both available at 12 and 24 months only).A key assumption in developing the PS estimand is the ‘monotonicity assumption’, which specified that no patient was ‘harmed’ by siponimod triggering relapses; that is, siponimod did not provoke relapses in patients who would not have relapsed with placebo (Figure 1). This assumption was assessed with sensitivity analyses.
Hypothetical estimands: disability progression independent of relapse effect in overall population
Two estimands were defined for these analyses, differing in the way in which on-study relapses were controlled: (1) population: patients in the EXPAND population defined by eligibility criteria; (2) variable: 3mCDP; (3) intercurrent event: confirmed on-study relapse using two strategies (first strategy: no patients would experience on-study relapses; second strategy: patients in both treatment arms have equal risk of experiencing on-study relapses); and (4) population-level summary: the hazard ratio.
Analysis of the hypothetical situation in which no patients would experience on-study relapses used a Cox proportional hazards model that censored patients at the time of first relapse. Baseline EDSS score and occurrence of relapses in the 2 years before randomization were covariates. Given that treatment influences timing of first relapse, there is a risk of bias by informative censoring. The inverse probability censoring weight (IPCW) was applied to account for this source of bias. 15
Analysis of the hypothetical situation in which patients in both treatment arms were equally likely to experience on-study relapses was based on a simulation approach from empirical distributions (a bootstrap-based method). Patients who relapsed on siponimod received increased weighting to compensate for relapses prevented by siponimod; thus, computed relapse rates in the simulated sample equal rates observed with placebo. These simulated studies were then analysed using a Cox proportional hazards model, with baseline EDSS score and occurrence of relapses in the 2 years before randomization as covariates. Limitations and key assumptions for the analyses undertaken are summarized in Table 1.
Exploratory analysis: frequency of no evidence of disease activity (NEDA-2) at month 12
The frequency with which patients achieved NEDA-2, defined as the absence of a confirmed relapse as well as of gadolinium-enhancing T1 lesions and new or enlarging T2 lesions prior to or at the MRI scan performed at month 12, is described using summary statistics.
Data availability
Raw data were generated at Novartis (Basel, Switzerland). Derived data supporting the study findings are available from the corresponding author on request.
Results
Patient characteristics
Of 1651 patients randomized in EXPAND, 1645 were included in the analysis. Of the six patients excluded, five never received study drug and one did not provide signed consent before commencing study procedures. 10 Overall, 36% (n = 590) of patients relapsed in the 2 years before randomization and 13% (n = 215) relapsed on study.
At randomization, patients without a pre-study relapse were older and had longer disease duration than those who had a pre-study relapse, although both groups had similar levels of baseline disability (based on EDSS score). In 18% (n = 189/1055) of patients without pre-study relapses, there was radiographic evidence of acute inflammatory activity at baseline (gadolinium-enhancing T1 lesions on brain MRI). Mean T2 lesion volume was similar in both groups (Table 2).
Disease characteristics at randomization and on-study clinical characteristics (relapses and annualized relapse rate) in patients with or without relapses in the 2 years before study enrolment.
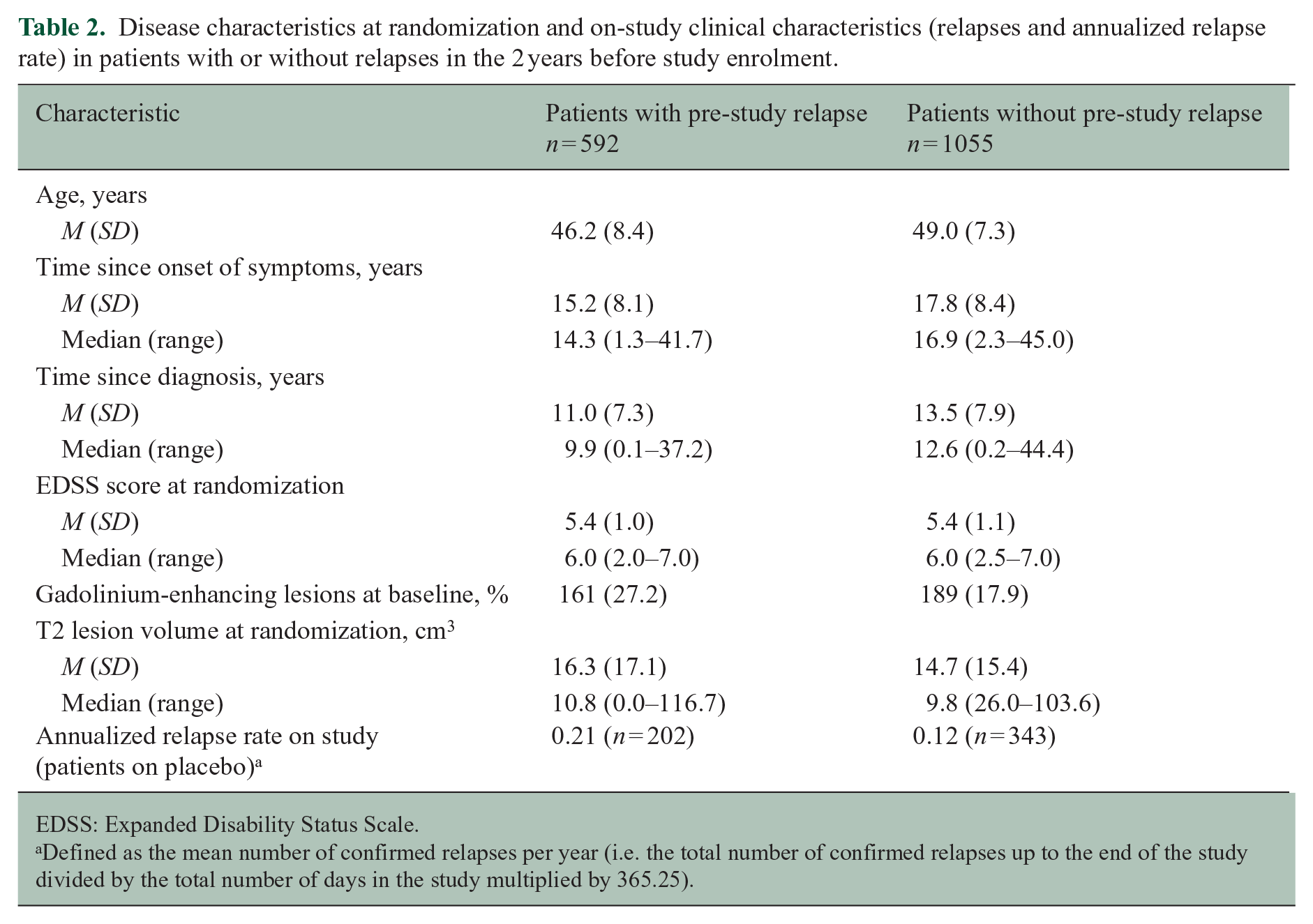
EDSS: Expanded Disability Status Scale.
Defined as the mean number of confirmed relapses per year (i.e. the total number of confirmed relapses up to the end of the study divided by the total number of days in the study multiplied by 365.25).
Traditional subgroup analyses
In patients with no relapses in the 2 years before enrolment, siponimod reduced risk of 3mCDP by 13% and risk of 6mCDP by 18% relative to placebo. 10 For on-study relapses, the risk reduction for 3mCDP was numerically greater in patients with on-study relapses compared to those without (20% vs 15%; Figure 2). For 6mCDP, the risk reduction was lower in patients with on-study relapses compared to those without (14% vs 24%; Figure 2). The only statistically significant reduction observed was for 6mCDP in the subgroup without on-study relapses. Of 343 placebo-treated patients with no relapses in the period before enrolment, 51 (14.9%) patients relapsed during the study.

Risk of 3mCDP and of 6mCDP with siponimod relative to placebo, by relapse-activity subgroup: Pre-specified analyses of 3mCDP and 6mCDP were undertaken in the subgroups of patients with or without on-study relapses. A Cox proportional hazards model was adjusted for treatment, country, relapses in the 2 years before study entry and EDSS score at randomization. 3mCDP: 3-month confirmed disability progression; 6mCDP: 6-month confirmed disability progression; EDSS: Expanded Disability Status Scale; HR: hazard ratio; n: number of patients with CDP; N: number of patients in the subgroup.
Principal stratum analysis
Depending on the exposure period evaluated, the PS model estimated that 80%–87% of patients were ‘non-relapsing’, consistent with the proportion of non-relapsing patients in the placebo group (81%). For context, the proportion of ‘definite-relapsing’ patients was in the range 8%–10% and of patients ‘benefitting’ in the range 5%–10% (Figure 3(a)). Across exposure periods, point estimates of siponimod’s effect on reducing 3mCDP ranged from 14%–20% and for 6mCDP ranged from 29%–33%, although this effect was statistically significant only for the 6mCDP outcome over a 12-month interval (Figure 3(b)).
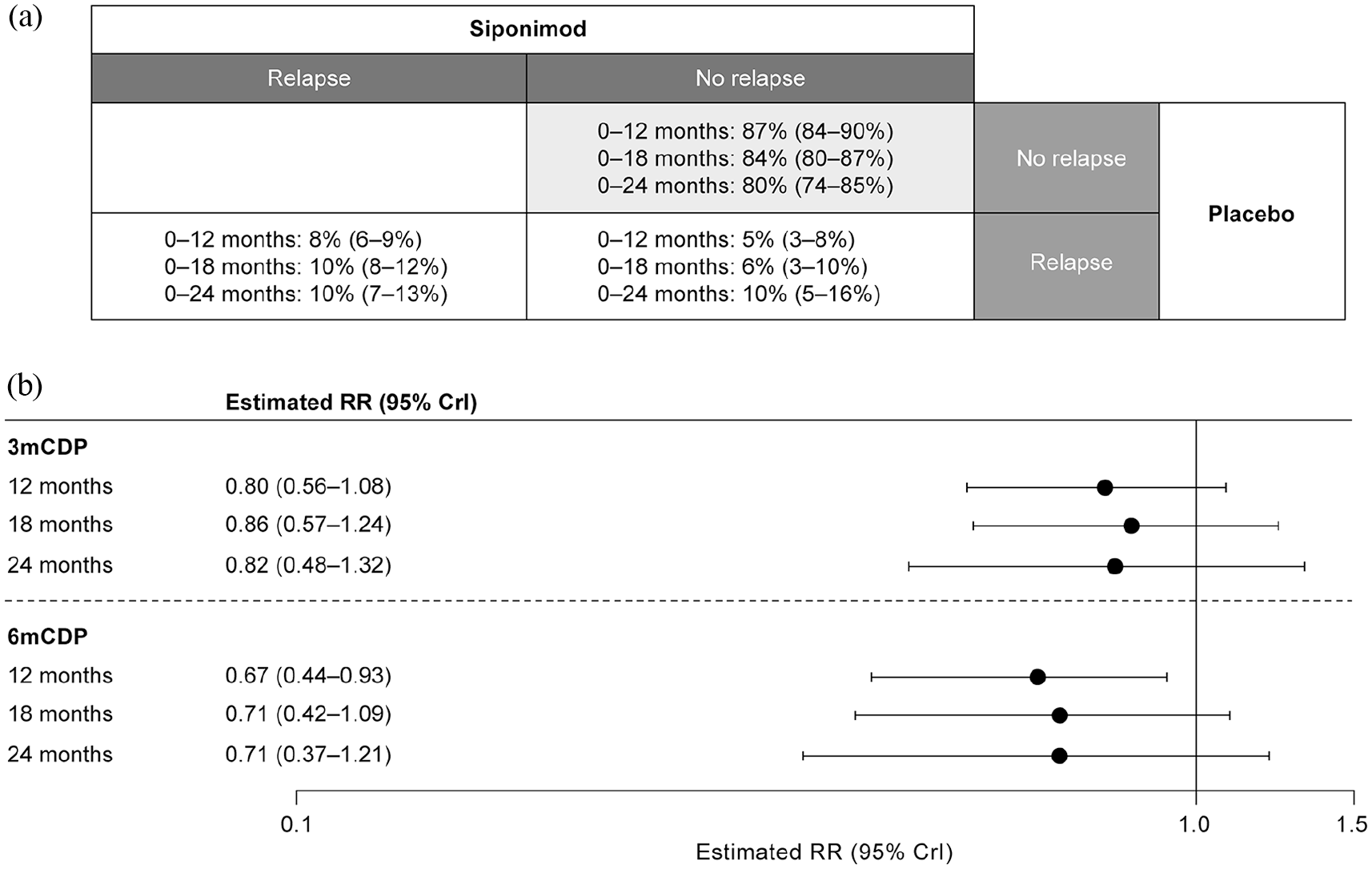
PS analysis of patients with confirmed relapses by time interval. (a) Proportion of patients by PS; (b) reduction in risk of 3mCDP and 6mCDP with siponimod compared with placebo in the ‘non-relapsing’ PS. In each case, the proportion and estimated RR are the median values of the respective posterior distribution, each shown with its associated 95% CrI. 3mCDP: 3-month confirmed disability progression; 6mCDP: 6-month confirmed disability progression; CrI: credible interval; PS: principal stratum; RR: risk ratio.
When unconfirmed relapses were included in the model, the estimated proportions of patients in each stratum (depending on the exposure period) were 75%–83% (‘non-relapsing’ PS), 11%–15% (‘definite-relapsing’), and 6%–10% (‘benefitting’; Figure 4(a)). In this model, siponimod reduced the risk of 3mCDP by 17%–20% and risk of 6mCDP by 29%–32% relative to placebo across exposure periods (Figure 4(b)).
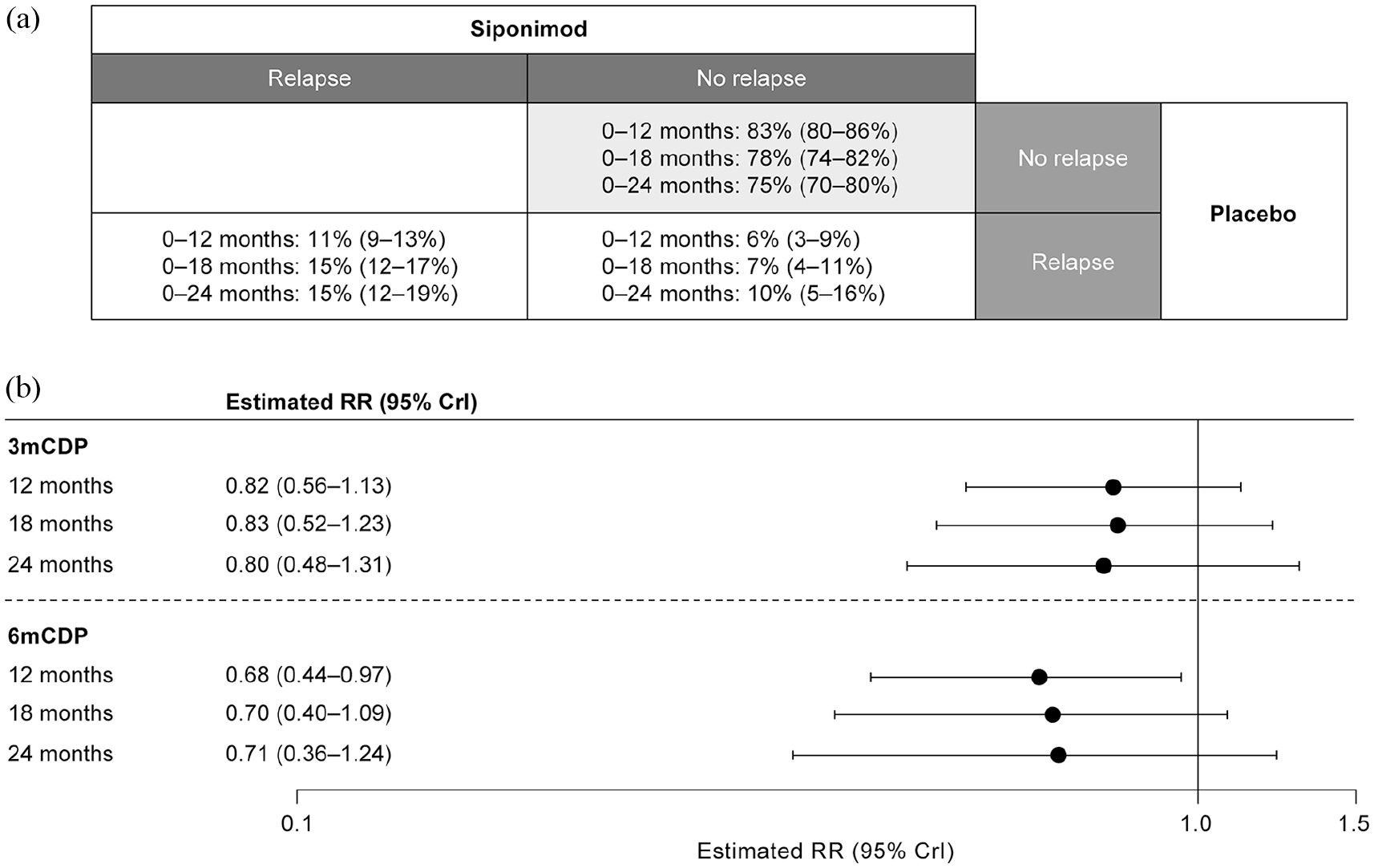
PS analysis of patients with confirmed and unconfirmed relapses by time interval. (a) Proportion of patients by PS; (b) reduction in risk of 3mCDP and 6mCDP with siponimod compared with placebo in the ‘non-relapsing’ PS. In each case, the proportion and estimated RR are the median value of the respective posterior distribution, shown with its associated 95% CrI. 3mCDP: 3-month confirmed disability progression; 6mCDP: 6-month confirmed disability progression; CrI: credible interval; PS: principal stratum; RR: risk ratio.
Modifying the model to include patients with no new or enlarging T2 lesions or those with fewer than two T2 lesions in the definition of the ‘non-relapsing’ stratum yielded results consistent with those obtained when the ‘non-relapsing’ stratum included only patients with no clinical relapses (Supplemental Table 1).
Sensitivity analyses testing the monotonicity assumption that no patients would be assigned to the harmed PS showed that this had little impact on the proportion of patients assigned to each stratum (Figure 5(a)) or on the degree to which siponimod reduced the risk of 3mCDP and 6mCDP compared with placebo in the ‘non-relapsing’ PS (Figure 5(b)).
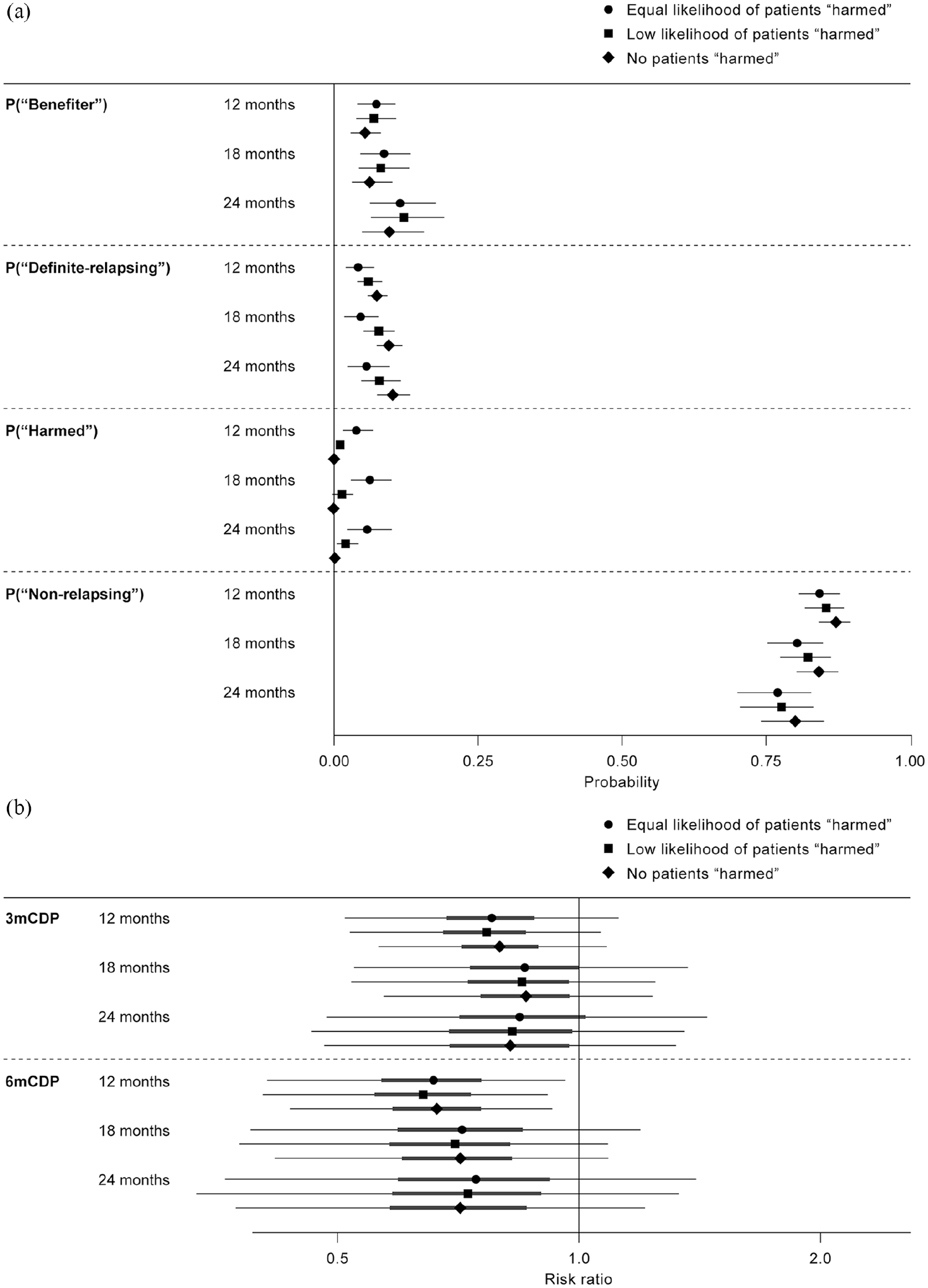
Sensitivity analysis of the assumption that no patients were classified as ‘harmed’. (a) Proportions of patients by stratum (95% credible interval); sensitivity analysis based on definitions of ‘harmed’ patients: two different assumptions were examined. The first assumption allowed for the possibility that some patients belonged to the ‘harmed’ stratum, but with a lower probability assigned to this scenario than to the other strata. The second assumption allowed for the possibility that the ‘harmed’ stratum was no larger or smaller than any other stratum. (b) PS analysis by time interval of reduction in risk of 3mCDP and 6mCDP with siponimod compared with placebo in the ‘non-relapsing’ PS; sensitivity analysis based on definitions of ‘harmed’ patients: posterior medians (point), and posterior 50% and 95% credible intervals (thick and thin lines, respectively) are shown.
Hypothetical scenarios: relapse-independent treatment effects in the overall population
Assuming no patients experienced on-study relapses, the estimated reduction in risk of 3mCDP with siponimod relative to placebo was 14% in the Cox model, with censoring at the time of first relapse and IPCW correction. For 6mCDP, risk reduction was 23% with IPCW correction; the estimate reaching nominal statistical significance (Figure 6). Assuming patients in both treatment arms had the same risk of experiencing on-study relapses, Cox regression analyses on samples simulated from empirical distribution yielded risk reductions with siponimod relative to placebo of 18% for 3mCDP and 23% for 6mCDP (Figure 6).
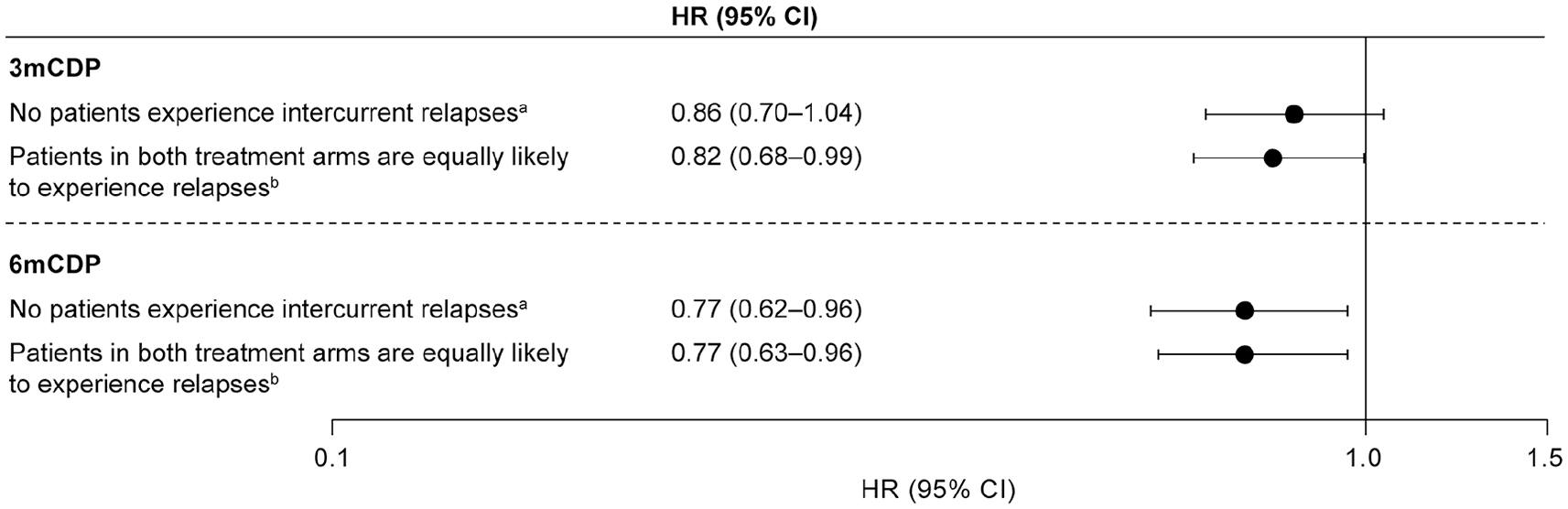
Hypothetical scenario: risk reduction in 3mCDP and 6mCDP with siponimod relative to placebo.
No evidence of disease activity (NEDA-2) at month 12
Overall, proportionally more patients achieved NEDA-2 with siponimod (56.9%; 517/909) than with placebo (42.2%; 184/436). In patients who achieved NEDA-2, proportionally fewer had 3mCDP with siponimod (17.8%; 92/517) than with placebo (21.2%; 39/184). The risk ratio was 0.84 (95% confidence interval (CI) = 0.60–1.17, p = 0.31, χ2 test); the between-group absolute risk difference was 3.4%. In patients who did not achieve NEDA-2, 22.4% (88/392) had 3mCDP with siponimod and 30.2% (76/252) with placebo. The risk ratio was 0.74 (95% CI = 0.57–0.97, p = 0.028); the between-group absolute risk difference was 7.7%
Discussion
To quantify the impact of siponimod on disability progression independent of relapse activity, we applied an estimand framework based on recommendations by regulatory bodies, 11 for use in situations in which on-study events, in this case clinical relapses, interfere with the interpretation of treatment effects (worsening disability). Our post hoc analyses showed that an important part of the benefit of siponimod on disability progression occurred independently of effects on relapse activity. In the analysis based on confirmed relapses, siponimod reduced the risk of 3mCDP by 14%–20% relative to placebo in the ‘non-relapsing’ PS, depending on the period of exposure analysed. This range is similar to the effect size seen in the overall population (risk reduction in time to 3mCDP, 21%). 10 Point estimates of the risk of 6mCDP in the ‘non-relapsing’ PS were 29%–33% lower with siponimod than placebo depending on the period of exposure, compared with a reduction in time to 6mCDP of 26% in the overall population. 10
While all three estimand methods yielded similar findings consistent with the primary endpoint and pre-planned analyses, certain assumptions were required: hypothetical scenarios assumed that either no relapses would occur or siponimod would have no effect on relapses. Both assumptions seem less plausible than those required for the PS analysis that focused on modelled patients who would not relapse at all within a given time frame.
Mediation analysis was previously applied in relapsing-remitting multiple sclerosis (RRMS) to address the question of surrogacy of MRI lesions and relapses for disability progression, 16 which raises the question of whether a similar approach (only focusing on the direct treatment effect) would be an appropriate analysis for our situation. However, our objective was to understand the effect of siponimod on disability progression in the absence of relapses (or in the absence of a treatment effect on relapses). The three estimands described above were considered as a way to address the question of interest more directly.
Participants who did not relapse on siponimod could have been assigned to either the ‘non-relapsing’ PS or the ‘benefitting’ PS. In the various exposure periods analysed, the ‘non-relapsing’ PS constituted 80%–87% of the analysed population, and it was assumed that these individuals would not relapse on either siponimod or placebo. The ‘benefitting’ PS was more stringent and assumed that patients would relapse on placebo but not on siponimod and, therefore, this stratum constituted a smaller group of 5%–10% of participants. This finding strongly suggests that the effect of siponimod on disability cannot be attributed only to its effect on relapses. A limitation of PS analysis is the monotonicity assumption, which requires that no participant would have relapsed on siponimod who would not have relapsed while on placebo. Sensitivity analyses that relaxed the monotonicity assumption (that no participants belonged in the ‘harmed’ PS) or that accounted for both unconfirmed and confirmed relapses had little impact on effect sizes in the ‘non-relapsing’ PS, confirming the robustness of the results.
There are several limitations that must be acknowledged. EXPAND was designed for the primary endpoint in the total population and not to prove a treatment effect of siponimod on disability progression independent of relapses. Furthermore, the event-driven design and variable follow-up times in EXPAND resulted in the number of analysable participants decreasing with longer follow-up periods. Consequently, the precision of the point estimates was reduced and credibility intervals were broadened. Indeed, credibility intervals for the PS analysis were considerably wider than in the two hypothetical estimand scenarios. This observation is attributable to the fact that the PS analysis considers subgroups based on a specific temporal interval, resulting in reduced sample size and loss of statistical power. In contrast, both hypothetical estimands target the full data set. Furthermore, only disability as measured by the EDSS was considered. The EDSS has several well-recognized limitations that are highly relevant to studying a progressive MS patient population and include dependence on ambulation and lack of sensitivity to major contributors to MS disability, including cognitive impairment and fatigue. Moreover, this study is a post hoc analysis and cannot be definitive.
Because EXPAND performed MRI scans annually, the potential contribution of new radiographic brain lesion formation to disability worsening is not readily interpretable. With annual MRI assessments, the stratum of participants in whom neither relapses nor new T2 lesions formed was too small to accurately assess a treatment effect. MRI scans would need to be performed at the same intervals as the disability assessments (every 3 months and at clinical relapses) and a substantially larger study size likely would be needed to have sufficient numbers of participants within the stratum of interest. Nonetheless, modifying the model to include participants with no new or enlarging T2 lesions yielded results that were pointing in the same direction as those obtained when only the absence of clinical relapses was used to define the non-relapsing patient stratum.
Regardless, an effect of lesion formation on progressive disability might not be identified. The radiographic burden of disease correlates poorly with disability: an observation referred to as the ‘clinico-radiological paradox’.17,18 Furthermore, progressive disability worsening seems to often occur independently of new lesion formation and commonly occurs independently of relapsing activity even in relapsing MS patients.19,20 Currently, the most robust MRI correlates of MS disability are spinal cord areas, whereas brain T2 lesion volumes correlate weakly with disability.21,22 Conventional MRI does not visualize microscopic, diffuse inflammatory processes that may underlie disease progression. Spinal cord imaging was not performed in the EXPAND trial and the contributions to disability progression of either spinal cord lesions and atrophy cannot be assessed in this data set.
In addition, clinical trials support the view that disability worsening in progressive forms of MS poorly correlated with new lesion formation. Despite showing robust effects on suppressing new lesion formation, the ASCEND clinical trial of natalizumab in SPMS failed to show an impact on disability. 3 A similar observation was found for fingolimod in primary progressive MS. 23 Many other treatments with robust effects on brain lesion formation in relapsing MS failed to reduce disability worsening in progressive MS. 7
As was seen in the overall EXPAND analysis, larger risk reductions in 6mCDP than in 3mCDP were observed. In general, 6mCDP is considered to be a more reliable measure of permanent disability progression than 3mCDP, which may be more affected by incomplete resolution of transient, relapse-related changes in disability.24,25 Thus, an effect on 6mCDP may be more meaningful in the context of progression independent of relapse. Although these analyses do not directly show that 3mCDP is influenced by relapses more than 6mCDP, the fact that reduction in risk of 6mCDP in the ‘non-relapsing’ PS was slightly greater than that seen in the overall population is consistent with this hypothesis.
Observations with siponimod in preclinical studies, including studies in which central effects manifest independently of effects in the peripheral immune system, lend biological plausibility to the clinical findings described here: siponimod readily crosses the blood–brain barrier 26 and can reduce demyelination and promote myelination.27,28 Siponimod also reduces grey matter inflammation, astrogliosis and microgliosis, independent of effects on peripheral lymphocytes. 29 The mechanisms underlying the central effects of siponimod in SPMS remain to be elucidated.
Primary analysis using the intention-to-treat principle corresponds to the ‘treatment policy’ estimand described in ICH E9 R1 11 and targets the treatment effect in all randomized patients regardless of relapses. This estimand is highly relevant, but cannot be used to estimate the treatment effect on disability independent of relapses. The convergence of findings across all pre-specified and post hoc analyses discussed here indicates that the effect of siponimod on disability is mostly independent of its effect on relapse activity, bolstering confidence in the utility of siponimod across the clinical spectrum of ambulatory patients with SPMS. To our knowledge, this is the first application of this estimand framework to any multiple sclerosis clinical trial.
This novel analytical approach provides a framework to resolve the long-standing problem of disentangling a treatment’s effect on relapses from that on disability progression. Prospectively implementing the approaches described here could be useful for distinguishing a therapy’s effects on relapse-associated disability from disability caused by progressive multiple sclerosis and should be considered by future studies where treatment may affect both relapses and disability progression independently. The outcomes of our analyses support that siponimod may be a useful treatment for SPMS, regardless of a patient’s relapse status.
Supplemental Material
Cree_EXPAND_CDP_Supplementary_materials_16Sep20 – Supplemental material for Siponimod: Disentangling disability and relapses in secondary progressive multiple sclerosis
Supplemental material, Cree_EXPAND_CDP_Supplementary_materials_16Sep20 for Siponimod: Disentangling disability and relapses in secondary progressive multiple sclerosis by Bruce AC Cree, Baldur Magnusson, Nicolas Rouyrre, Robert J Fox, Gavin Giovannoni, Patrick Vermersch, Amit Bar-Or, Ralf Gold, Daniela Piani Meier, Göril Karlsson, Davorka Tomic, Christian Wolf, Frank Dahlke and Ludwig Kappos in Multiple Sclerosis Journal
Footnotes
Acknowledgements
Medical writing assistance was provided by Kim Wager (Oxford PharmaGenesis, Oxford, UK), with input from all authors. Oxford PharmaGenesis, Oxford, UK, copyedited and styled the manuscript as per the journal requirements. Medical writing and editorial services were funded by Novartis Pharma AG.
Declaration of Conflicting Interests
The author(s) declared the following potential conflicts of interest with respect to the research, authorship, and/or publication of this article: B.A.C.C. has received personal compensation for consulting from Akili, Alexion, Atara, Biogen, EMD Serono, Sanofi, Novartis – which owns patent rights to siponimod that was used in this study, and TG Therapeutics. B.M. is an employee of Novartis Pharma AG, which owns patent rights to siponimod that was used in this study. N.R. is an employee of Novartis Pharma AG, which owns patent rights to siponimod that was used in this study. R.J.F. reports personal fees from Actelion, Biogen, EMD Serono, Genentech, Novartis – which owns patent rights to siponimod that was used in this study, and Teva; grants from Novartis; and other support from Biogen (clinical trial contracts). G.G. reports personal fees for consultancy from AbbVie (steering committee: daclizumab trials), Eisai, Elan, Five Prime, Genentech, GlaxoSmithKline, GW Pharmaceuticals, Pfizer and Synthon BV; grants from UCB Pharma; grants and personal fees for consultancy from Bayer-Schering, Biogen (steering committee: BG12 and daclizumab trials), Canbex, Genzyme/Sanofi, Ironwood, Merck Serono, Novartis – which owns patent rights to siponimod that was used in this study (steering committee: fingolimod and siponimod trials), Roche (steering committee: ocrelizumab trials) and Teva (steering committee: laquinimod trials); and fees for speaking at physicians’ summits and several medical education meetings from agencies or sponsoring companies. He is also the Co-Chief Editor of Multiple Sclerosis and Related Disorders (Elsevier). P.V. reports personal fees from Bayer HealthCare, Biogen Idec, Celgene, Merck Serono, Novartis – which owns patent rights to siponimod that was used in this study, Roche, Sanofi, Servier and Teva Neuroscience, and he or the institution he works for has received research support from Bayer HealthCare, Biogen Idec, Merck Serono, Novartis and Teva Neuroscience; he has also received fees as a journal editor from SAGE and Thieme Verlag. A.B.-O. has received personal compensation for consulting, serving on scientific advisory boards or speaking activities, or a combination thereof, from Atara Biotherapeutics, Bayer, Bayhill Therapeutics, Berlex, Biogen Idec, BioMS, Brainstorm, Celgene/Receptos, DioGenix, Eli Lilly, F Hoffmann-La Roche, Genentech, GlaxoSmithKline, Guthy-Jackson/GGF, Mappi Pharma, Medimmune, Merck Serono, Novartis – which owns patent rights to siponimod that was used in this study, Ono Pharmacia, Roche, Sanofi-Aventis, Teva Neuroscience and Wyeth. R.G. has received compensation for serving as a consultant or speaker from Bayer HealthCare, Biogen Idec, Merck Serono, Novartis and Teva Neuroscience, and he or the institution he works for has received research support from Bayer HealthCare, Biogen Idec, Merck Serono, Novartis and Teva Neuroscience; he has also received fees as a journal editor from SAGE and Thieme Verlag. D.P.M. is an employee of Novartis Pharma AG, which owns patent rights to siponimod that was used in this study. G.K. is an employee of Novartis Pharma AG, which owns patent rights to siponimod that was used in this study. D.T. is an employee of Novartis Pharma AG, which owns patent rights to siponimod that was used in this study. C.W. is a partner at Lycalis sprl. His institution has received fees from Celgene, Desitin, ICON, Mylan, Novartis, which owns patent rights to siponimod that was used in this study, Synthon and Teva. F.D. is an employee of Novartis Pharma AG, which owns patent rights to siponimod that was used in this study. L.K. reports that, in the past 3 years, his institution (University Hospital, University of Basel) has received funding used exclusively for research support and educational activities from Actelion, Alkermes, Allergan, Almirall, Bayer, Biogen, Celgene, CSL Behring, df-mp, the European Union, EXCEMED, GeNeuro, Genzyme, Japan Tobacco, Innoswiss, the Swiss Multiple Sclerosis Society, Merck, Mitsubishi Pharma, Minoryx, Novartis – which owns patent rights to siponimod that was used in this study, Pfizer, Receptos/Celgene, Roche, Roche Research Foundations, Sanofi-Aventis, Santhera, the Swiss National Research Foundation, Teva, UCB Pharma and Vianex, and licence fees for Neurostatus products.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: Novartis Pharma AG funded the study, data analyses and medical writing support.
Supplemental Material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.