
Abstract
Background:
The double-blind TERIKIDS study demonstrated the efficacy and safety of teriflunomide.
Objective:
To evaluate the efficacy, safety, and tolerability of continuous teriflunomide treatment in the TERIKIDS open-label extension.
Methods:
In the double-blind period, children with relapsing MS were randomized to placebo or teriflunomide (14 mg adult-equivalent dose) for ⩽ 96 weeks. Participants received teriflunomide for ⩽ 192 weeks post-randomization in the open-label extension.
Results:
The mean age at screening was 14.6 years. For teriflunomide/teriflunomide versus placebo/teriflunomide, estimated clinical relapse risk was reduced by 38% (hazard ratio (HR) 0.62; 95% confidence interval (CI) 0.39–0.98; p = 0.11) and numbers of gadolinium-enhancing T1 and new/enlarging T2 lesions were reduced by 43% (relative risk (RR) 0.570; 95% CI 0.33–0.98; p = 0.043) and 49% (RR 0.511; 95% CI 0.34–0.76; p = 0.001), respectively, in the combined double-blind and open-label periods. There was a trend toward reduced risk of 24-week sustained disability progression for teriflunomide/teriflunomide versus placebo/teriflunomide (HR 0.47; 95% CI 0.23–0.96). During the open-label extension, incidences of safety-related discontinuations were 4.0% (teriflunomide/teriflunomide) and 13.5% (placebo/teriflunomide), including two children who developed pancreatitis in the teriflunomide/teriflunomide group.
Conclusion:
Teriflunomide reduced the long-term risk of focal inflammatory activity, with generally manageable tolerability and no new safety signals. Further evidence would strengthen clinical efficacy findings.
ClinicalTrials.gov: NCT02201108.
Introduction
Pediatric multiple sclerosis (MS), commonly defined as MS onset before 18 years of age, represents 2%–5% of all people with MS.1–4 An estimated 98% of children with MS have a relapsing-remitting course at onset, compared with 84% of adults with MS. 5 Children with MS also typically have higher relapse rates 6 and a greater burden of lesions on magnetic resonance imaging (MRI), compared with adults.1,7 Pediatric-onset MS is characterized by a slower rate of permanent neurological disability accrual compared with adult-onset disease.5,8,9 Nevertheless, irreversible disability landmarks have reached a median of 10 years earlier in pediatric-onset versus adult-onset MS. 5 Similar genetic and environmental risk factors for MS are present in pediatric-onset MS and adult-onset MS, implicating a similar underlying pathophysiology regardless of the age at onset. 10
Despite the recognized prevalence and risk of early adulthood disability for children with MS, approved treatment options are limited, and randomized clinical trials have been, until recently, relatively scarce.11–15 Observational data derived from off-label use of treatments approved for adults with MS indicates that the efficacy and safety profiles of several disease-modifying therapies may be similar in children and adults with MS. 16 Teriflunomide, approved in adults with relapsing MS, has shown efficacy in clinical studies of adults17–19 with a well-characterized and manageable safety profile that has remained consis-tent with up to 12 years of exposure.20–22
In the 2-year, randomized, double-blind, phase 3 TERIKIDS study (NCT02201108) of pediatric participants with relapsing MS, teriflunomide reduced the combined risk of clinical relapse or high MRI activity compared with placebo (hazard ratio (HR) 0.57; 95% confidence interval (CI) 0.37–0.87; p = 0.04). The relative reduction in the adjusted number of gadolinium (Gd)-enhancing T1 and new/enlarging T2 lesions versus placebo was 75% (p = 0.0001) and 55% (p = 0.00061), respectively. The safety profile was generally favorable, although two children treated with teriflunomide on the study experienced pancreatitis necessitating treatment discontinuation (both participants recovered). 15 On the basis of the TERIKIDS study findings, the approved indication for teriflunomide in the European Union has been extended to include the treatment of children and adolescents aged 10 years and older with relapsing-remitting MS. 23
Here, we report the safety and efficacy outcomes from up to 2 years of additional follow-up in the open-label, long-term extension of the TERIKIDS study.
Materials and methods
Study design and participants
TERIKIDS was a multicenter, phase 3, double-blind, randomized, placebo-controlled, parallel-group study consisting of a screening period of up to 4 weeks, a double-blind core treatment period of up to 96 weeks, and an optional open-label extension period of up to 192 weeks after randomization (see online Supplemental Figure 1). The study was conducted in 22 countries across Asia, Europe, the Middle East, North Africa, and North America. Participants 10–17 years of age who fulfilled the consensus definition for pediatric MS and who had one or more relapses in the 12 months preceding screening or ⩾ 2 relapses in the 24 months preceding screening were enrolled. Key exclusion criteria were an Expanded Disability Status Scale (EDSS) score > 5.5 at screening or during randomization, relapse within 30 days before randomization, and weight < 20 kg. See online Supplemental Table 1 for full inclusion/exclusion criteria.
All participants underwent an 8-week run-in phase at the beginning of the double-blind period and were randomized 2:1 to the adult-equivalent oral 7 mg dose based on body weight (⩽40 kg, 3.5 mg teriflunomide; >40 kg, 7 mg teriflunomide) or matching placebo. At the end of the run-in, the teriflunomide dose was adjusted and maintained for the remainder of the study to ensure exposure similar to the once-daily adult-equivalent dose of teriflunomide 14 mg.
Participants who completed the 96-week core double-blind period or qualified for early switch to open-label teriflunomide could continue in the open-label extension until 192 weeks after initial randomization. Criteria for early switch to open-label teriflunomide consisted of an adjudicated relapse or evidence of high MRI activity, defined as ⩾9 new/enlarged T2 lesions at week 36 and ⩾5 new/enlarged T2 lesions on each of 2 consecutive MRI scans at weeks 36 and 48 or at weeks 48 and 72. All participants entering the open-label extension from the placebo arm or teriflunomide arm underwent a second run-in phase of 8 weeks and were followed through 192 weeks. Participants ran-domized to placebo in the core study started the open-label extension on the once-daily teriflunomide 7 mg adult-equivalent dose, which was adjusted to the adult-equivalent 14 mg dose, and participants initially ran-domized to teriflunomide continued on the same adjusted dose administered in the core phase. In the open-label extension, participants initially randomized to teriflunomide are referred to as the teriflunomide/teriflunomide group, and those randomized to placebo in the double-blind period are referred to as the placebo/teriflunomide group. All participants in the open-label extension received teriflunomide at the body weight-based 14 mg adult-equivalent dose.
The TERIKIDS study protocol was approved by an institutional review board or independent ethics committee as per local regulations, and the study was conducted in accordance with the Declaration of Helsinki, the World Medical Assemblies, and the International Council for Harmonization guidelines for Good Clinical Practice. Written informed consent was obtained from all participants and their parents or legal guardians according to local regulations.
Study endpoints
The primary endpoint was the time to first clinical relapse after randomization in the combined double-blind and open-label extension periods. Clinical relapses, confirmed by an independent adjudication panel, were defined as new or recurrent neurological symptoms lasting ⩾ 24 hours that were not associated with fever or infection and were accompanied by new objective neurological findings on examination. Key secondary endpoints were MRI disease activity (defined as the number of Gd-enhancing T1 lesions and the number of new/newly enlarged T2 lesions per MRI scan during the combined double-blind and open-label extension periods), the time to sustained disability progression on the EDSS (defined as a sustained increase of ⩾ 1.0 point on the EDSS or 0.5 points for participants with baseline EDSS > 5.5 persisting for ⩾ 24 weeks from baseline EDSS during the combined double-blind and open-label extension periods), and the safety and tolerability of teriflunomide during the open-label extension based on physical examinations, vital signs, laboratory assessments, and adverse event reporting.
Statistical analysis
Details about the statistical analysis plan for the core double-blind treatment period are found in Supplemental Table 2. Efficacy and safety data were assessed for all participants entering the open-label extension.
Results
Participant disposition
A total of 152 (91.2%) of the 166 participants randomized in the core double-blind period enrolled in the open-label extension, including 100 from the teriflunomide group (91.7%) and 52 from the placebo group (91.2%) (Figure 1). Of the 104 (68.4%) participants who completed the open-label extension, 73 participants were originally randomized to teriflunomide (73.0%), and 31 were from the placebo arm (59.6%). Lack of efficacy and adverse events were the most common reasons for treatment discontinuation in both groups.
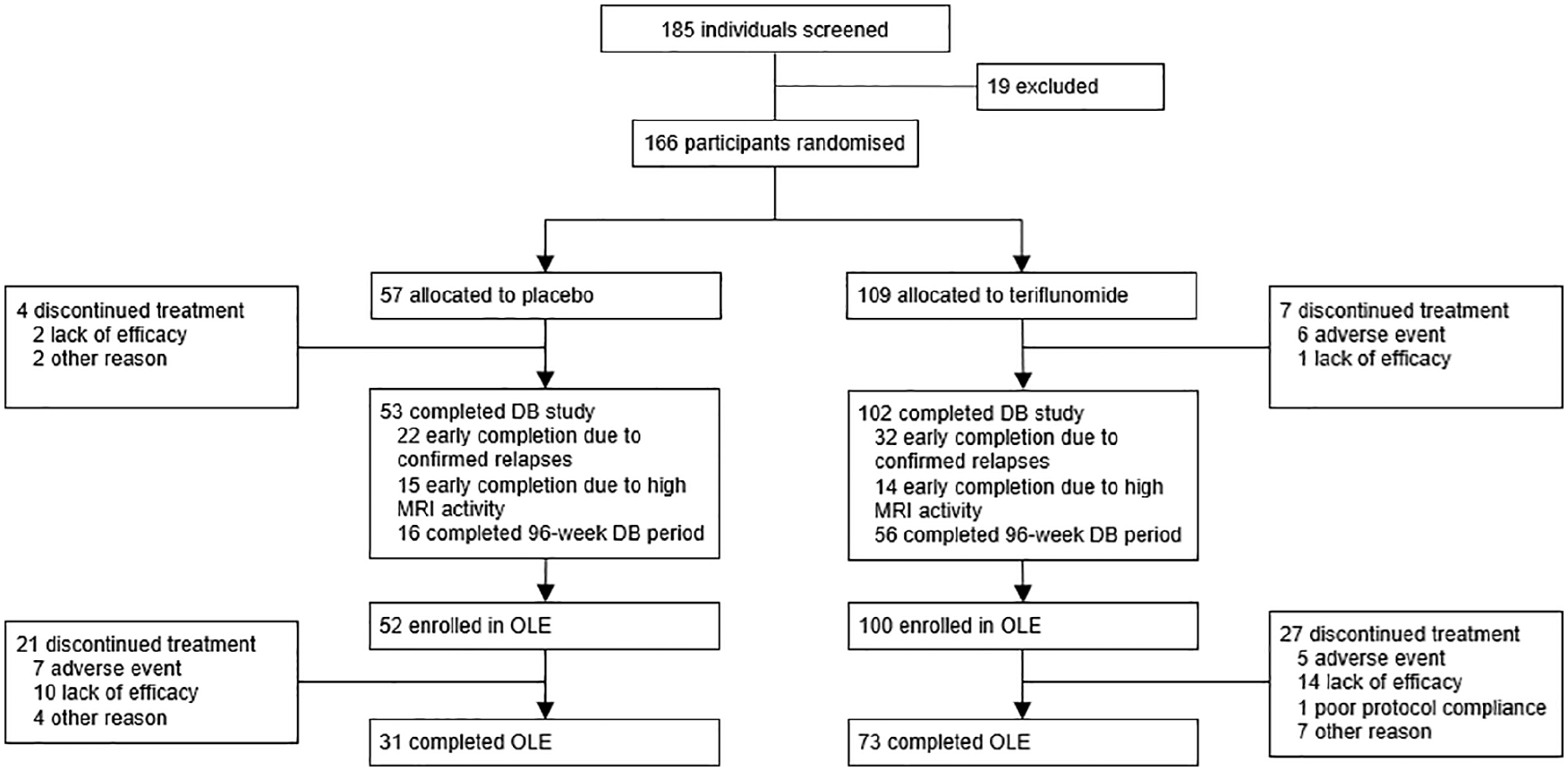
TERIKIDS participant disposition.
Baseline demographic and disease characteristics
Demographic and disease characteristics were recorded at baseline in the core double-blind period and were generally well-balanced between groups (Table 1). The mean age at screening for the double-blind study in both groups was 14.6 years. In the teriflunomide/teriflunomide group, 85.0% of participants were 13 years old or older at baseline (versus 80.8% in the placebo/teriflunomide group) and had a correspondingly higher Tanner Scale pubertal status. The mean time since MS diagnosis was approximately 1.5 years, and the mean time from the onset of MS symptoms was approximately 2.4 years. The mean (SD) number of relapses in the 2 years before entering the core double-blind period was 2.1 (1.0). Prior MS treatments were received by 18.0% of participants in the teriflunomide/teriflunomide group and 26.9% in the placebo/teriflunomide group.
Baseline demographic and disease characteristics. a
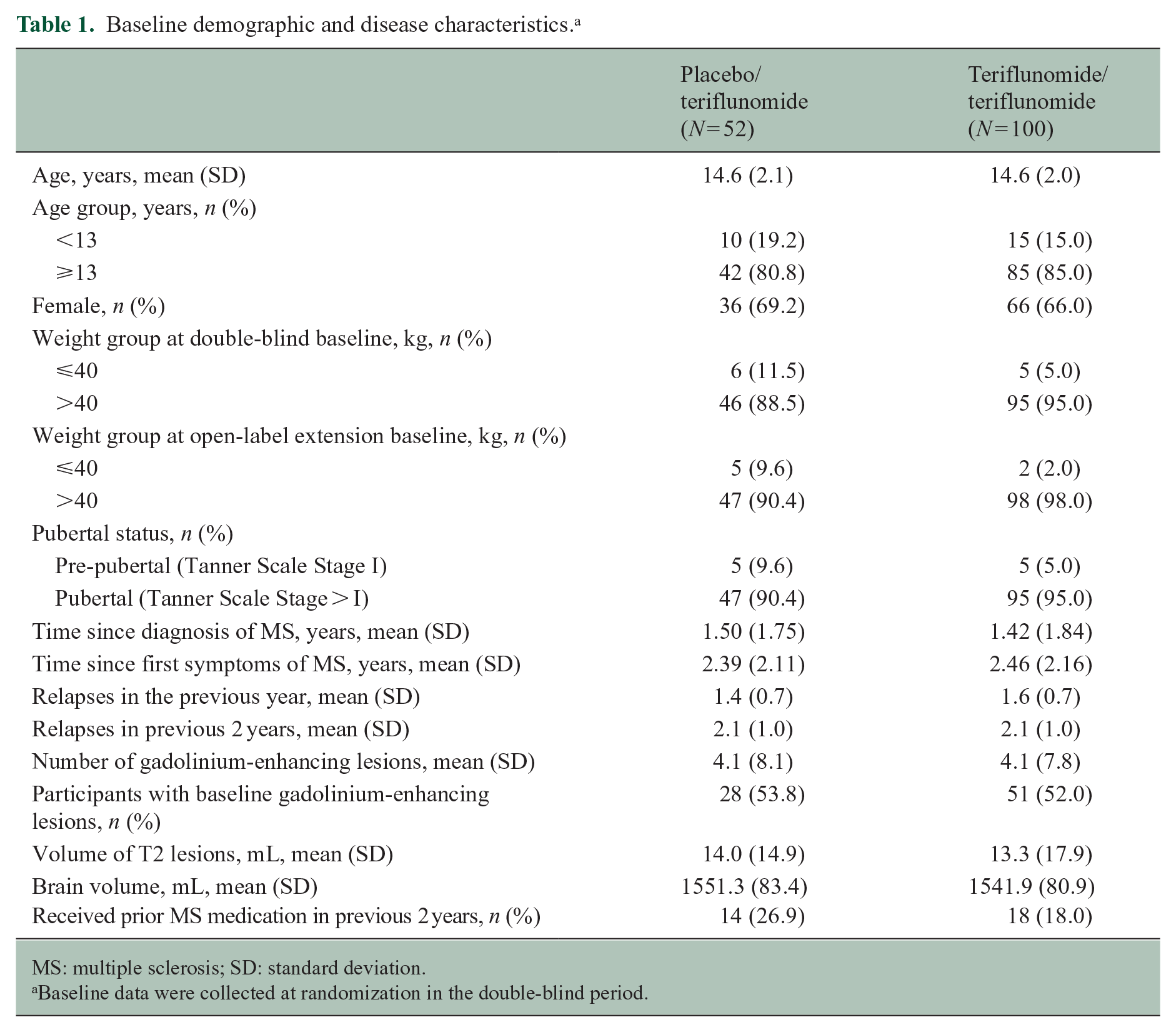
MS: multiple sclerosis; SD: standard deviation.
Baseline data were collected at randomization in the double-blind period.
Efficacy
The estimated risk of clinical relapse from randomization in the double-blind treatment period through the open-label extension using the Kaplan-Meier method was reduced by 38% (HR 0.62; 95% CI 0.39–0.98; p = 0.11) for participants in the teriflunomide/teriflunomide group compared with the placebo/teri-flunomide group (Figure 2). The corresponding median times to the first on-study clinical relapse were longer for the teriflunomide/teriflunomide group (153.9 weeks) than for the placebo/teriflunomide group (86.0 weeks).
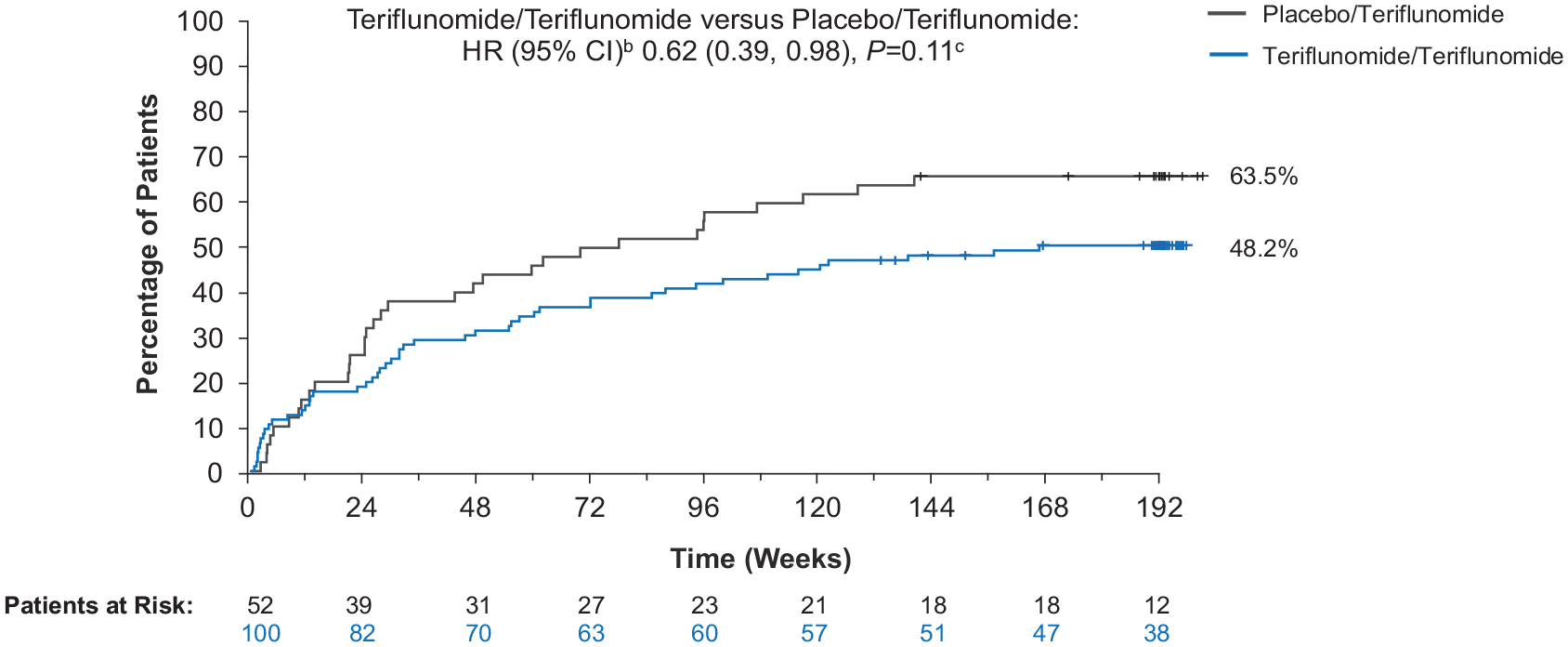
Time to first clinical relapsea, combined double-blind and open-label extension periods.
Participants in the teriflunomide/teriflunomide group had fewer Gd-enhancing T1 lesions (1.5 versus 2.7) and new/enlarging T2 lesions (5.7 versus 11.1) compared with participants in the placebo/teriflunomide group, respectively (Figure 3). The number of Gd-enhancing T1 lesions was reduced by 43% (relative risk (RR) 0.570; 95% CI 0.33–0.98; p = 0.043) in the teriflunomide/teriflunomide group compared with the placebo/teriflunomide group. Similarly, the number of new/enlarging T2 lesions was reduced by 49% (RR 0.511; 95% CI 0.34–0.76; p = 0.001) in the teri-flunomide/teriflunomide group versus the placebo/teriflunomide group.
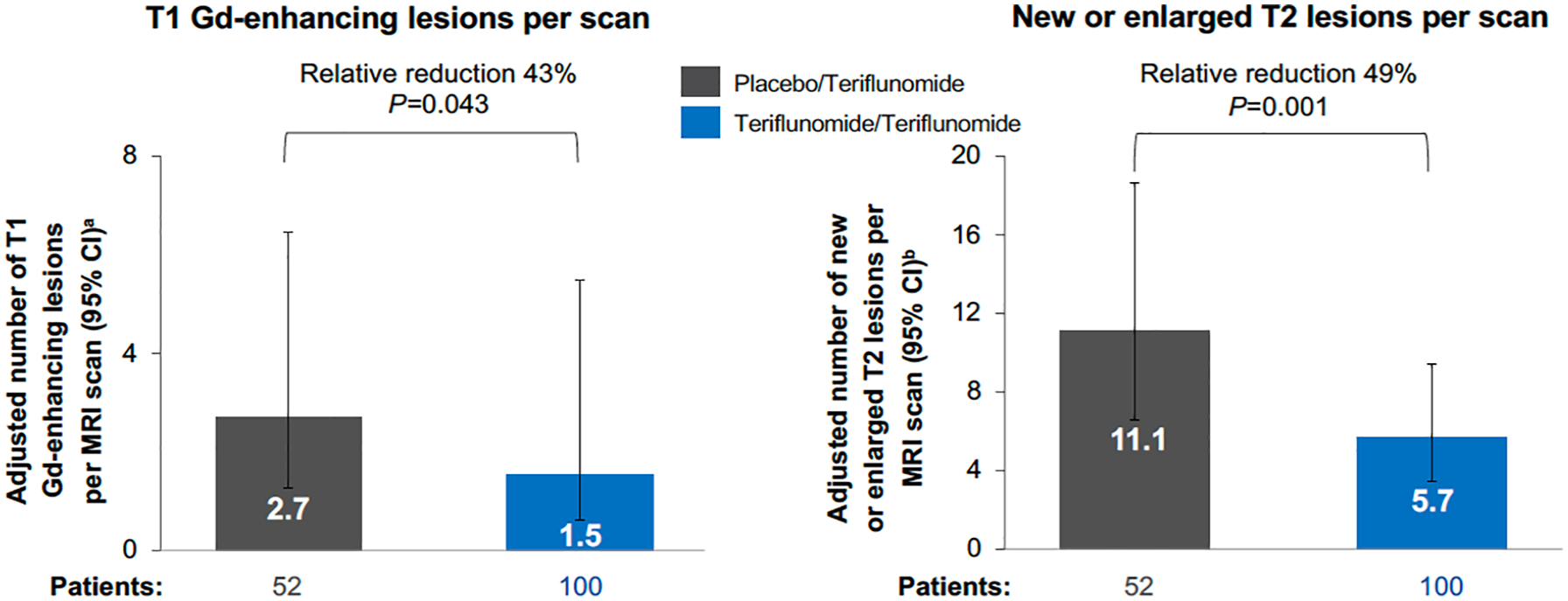
MRI outcomes, combined double-blind and open-label extension periods.
The estimated risk of disability progression sustained for 24 weeks at Week 192 using the Kaplan-Meier method was 0.163 (95% CI 0.096–0.246) in the teriflunomide/teriflunomide group and 0.323 (95% CI 0.188–0.466) in the placebo/teriflunomide group. The risk of disability progression sustained for 24 weeks was numerically lower in the teriflunomide/teriflunomide group compared with the placebo/teriflunomide group (HR 0.465; 95% CI 0.226–0.959) (Figure 4). EDSS scores were generally stable over the combined core double-blind and extension periods; median (interquartile range (IQR)) EDSS score was 1.5 (1.0–2.0) at baseline and 1.5 (0.0–2.0) at Week 192 for teriflunomide/teriflunomide and 1.5 (1.0–2.0) and 1.5 (0.0–3.0) for placebo/teriflunomide (Supplemental Figure 2). The majority of participants had an EDSS score < 2 at baseline (81% and 67% for teriflunomide/teriflunomide and placebo/teriflunomide, respectively) and during treatment (66%–76% and 53%–71%, respectively). Fewer than 4% of participants in the teriflunomide/teriflunomide group and 8% in the placebo/teriflunomide group had an EDSS score > 4 (Supplemental Figure 3). The EDSS scores were relatively low, indicating near-normal neurological examinations.
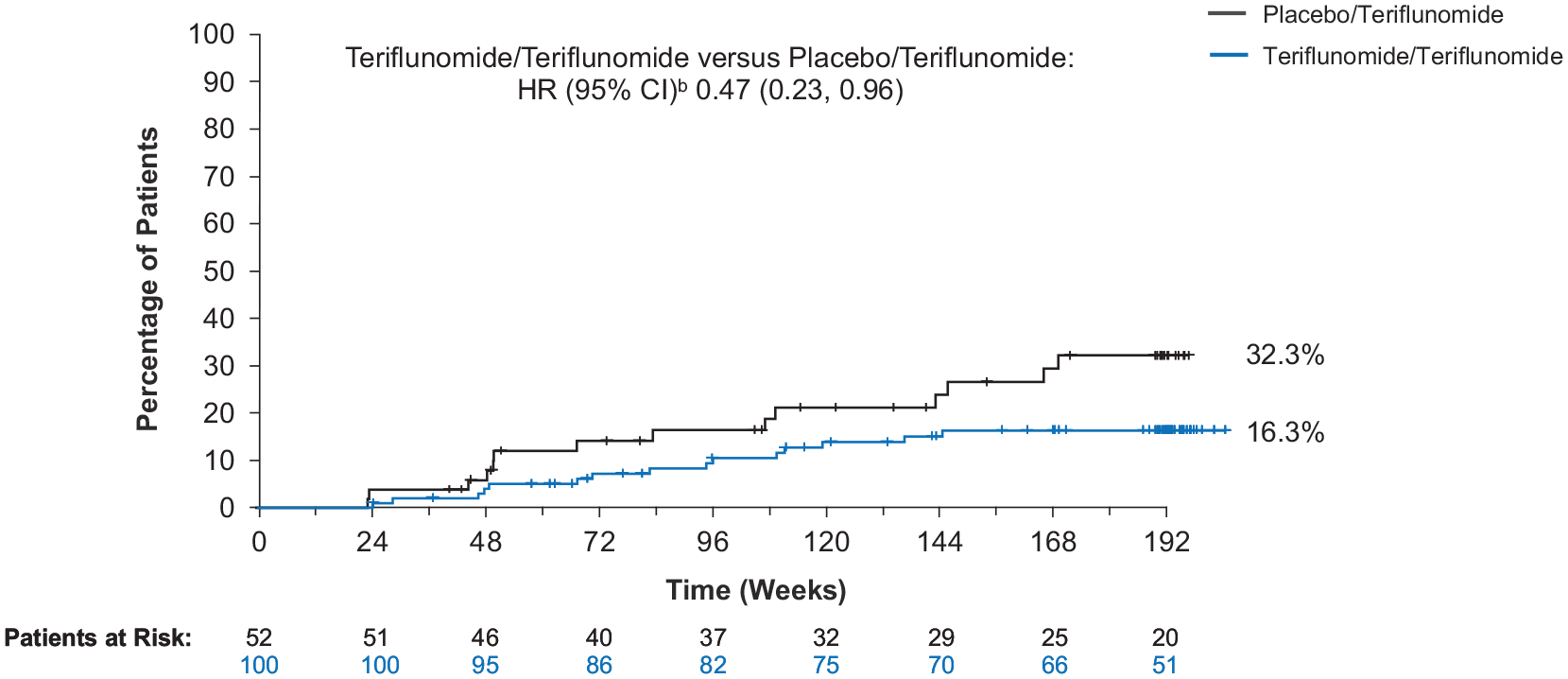
Time to disability progression sustained for 24 weeks, combined double-blind and open-label extension periods.a
Safety
Overall, teriflunomide was well tolerated with a manageable safety profile, generally consistent with the results observed in the double-blind period. 15 Compared with the placebo/teriflunomide group, participants in the teriflunomide/teriflunomide group had a lower incidence of treatment-emergent adverse events (TEAEs; 90.4% vs. 81.0%) and serious TEAEs (28.8% vs. 14.0%) during the open-label extension (Table 2). TEAEs occurring in ⩾ 10.0% of participants in the teriflunomide/teriflunomide group or the placebo/teriflunomide group were nasopharyngitis, headache, dizziness, alopecia, increased alanine aminotransferase, and accidental overdose of teriflunomide (2 doses in less than 24 hours). Of the 14 accidental overdose events occurring in 12 participants (7.9%), 12 events were mild in nature and 2 were moderate. All events were resolved without corrective treatment. In addition, two participants intentionally took an overdose of teriflunomide, including a participant who took 18 tablets in an attempted suicide related to a failure at school. The participant experienced sensory disorders and tingling sensations transiently for less than 2 hours, was hospitalized for 12 hours, and recovered following temporary teriflunomide discontinuation and accelerated elimination.
TERIKIDS safety outcomes in the open-label extension.
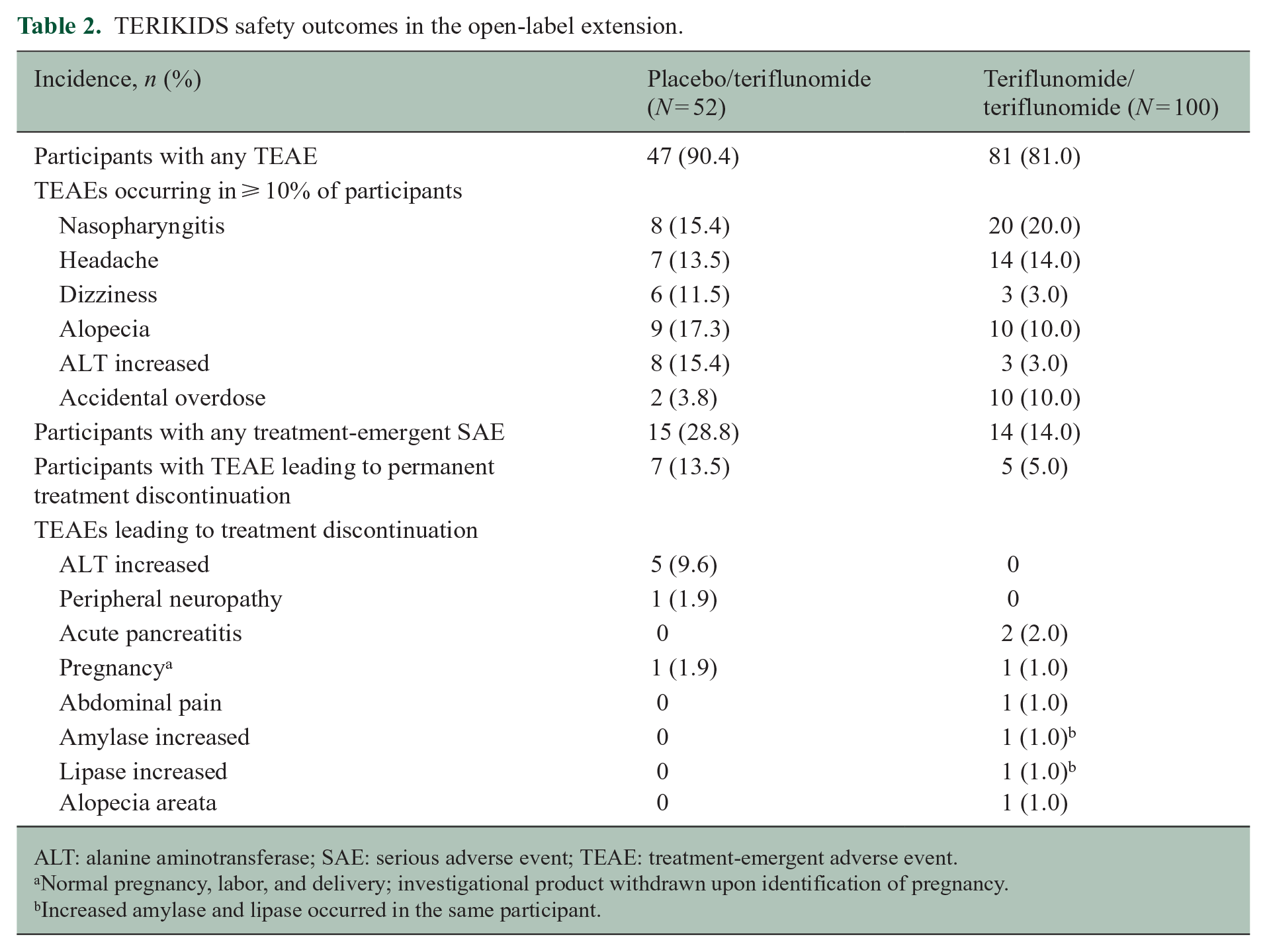
ALT: alanine aminotransferase; SAE: serious adverse event; TEAE: treatment-emergent adverse event.
Normal pregnancy, labor, and delivery; investigational product withdrawn upon identification of pregnancy.
Increased amylase and lipase occurred in the same participant.
Permanent discontinuation of treatment due to TEAEs during the open-label extension occurred in 5.0% of participants in the teriflunomide/teriflunomide group and 13.5% of participants in the placebo/teriflunomide group. Twelve participants in both groups discontinued open-label treatment due to TEAEs, including two cases of acute pancreatitis in the teriflunomide/teriflunomide group (Table 2). In the first case of acute pancreatitis, reported previously, 15 the participant experienced lipase increase (16.15 × upper limit of normal) after 1 year of teriflunomide treatment. Abdominal imaging identified acute pancreatitis with pancreatic nodules and inflammatory lesions of the pancreas. After treatment discontinuation, lipase levels normalized and the participant recovered. In the second case, not reported previously, the participant experienced abdominal pain with lipase and amylase increase after approximately 3 years of teriflunomide treatment. Abdominal imaging identified radiological signs of pancreatitis. The participant recovered after discontinuation of treatment.
The TEAEs occurring in ⩾ 10.0% of all participants from randomization through the end of the open-label extension were nasopharyngitis (26.7%), alopecia (23.0%), upper respiratory tract infection (23.0%), headache (19.9%), diarrhea (12.4%), influenza (11.8%), abdominal pain (11.2%), and dizziness (9.9%). Most TEAEs were mild to moderate in severity. Of the 19 alopecia events, 16 were mild, 2 were moderate, and 1 was severe, with most cases recovering during the study period. Severe TEAEs occurred in 15.5% of participants; most individual severe TEAEs were reported in < 1% of participants with the exception of increased blood creatinine phosphokinase (2.5%), headache (1.2%), and acute pancreatitis (1.2%).
Discussion
The long-term results of the TERIKIDS study open-label extension show that teriflunomide treatment for up to 192 weeks has long-term beneficial effects for children with relapsing MS. Continuous teriflunomide reduced the risk of clinical relapse by 38% compared with delayed teriflunomide (p = 0.11), with a corresponding longer median time to first clinical relapse (153.9 versus 86.0 weeks, respectively). Statistically significant benefits were observed in MRI activity for participants in the continuous teriflunomide group compared with delayed teriflunomide, with a 43% reduction in the number of Gd-enhancing T1 lesions (p = 0.043) and a 49% reduction in the number of new/enlarging T2 lesions (p = 0.001). The risk of 24-week sustained disability progression was decreased by 53% for pediatric participants receiving continuous teriflunomide compared with delayed teriflunomide; however, this result should be interpreted with caution since most participants had EDSS scores less than 2, indicating minimal disability.
The treatment effect of teriflunomide in TERIKIDS was similar to the clinical relapse and MRI findings in adults with relapsing MS from the phase 3 TEMSO 17 and TOWER 18 studies, a phase 2 placebo-controlled, 6-month study 24 and its core plus extension study of up to 8.5 years, 21 and a retrospective analysis of older people with relapsing MS who switched from other disease-modifying therapies to teriflunomide. 25 The efficacy of teriflunomide in children with relapsing MS is consistent with the apparent age-independence of teriflunomide’s clinical efficacy suggested by a pre-specified sub-group analysis of younger (<38 years) and older adults (⩾38 years) in TEMSO. 26 The treatment effect estimate for the time to first clinical relapse in the present analysis of the combined double-blind and open-label periods (HR 0.62; 95% CI 0.39–0.98) was very similar to the corresponding estimate from the analysis of the double-blind period (HR 0.66; 95% CI 0.39–1.11). 15 However, caution should be applied when interpreting the related P-values derived from these analyses since the analysis of the double-blind period censored participants who switched to open-label teriflunomide treatment early as a result of high MRI activity, whereas the combined period analysis included all data regardless of rescue therapy. In the analysis of the double-blind period, a relatively high number of participants in the placebo group were censored due to MRI activity compared with the teriflunomide group (26.3% vs. 12.8%), which reduced the power of this analysis and probably biased the result against teriflunomide. 15
Overall, the tolerability of teriflunomide was generally manageable in this pediatric population followed for up to 4 years, and the adverse event profile in the open-label extension was similar to the double-blind period. 15 Rates of treatment discontinuation due to TEAEs were 4.0% and 13.5% for the teriflunomide/teriflunomide and placebo/teriflunomide groups, respectively, which is similar to that observed in the clinical studies in adults.17,18,21,24 The most frequently reported TEAEs in TERIKIDS, including nasopharyngitis, alopecia, upper respiratory tract infection, headache, and diarrhea, were also most commonly observed in adults treated with teriflunomide 14 mg.17,20,24 The higher incidence of increased alanine aminotransferase and alopecia in the placebo/teriflunomide group compared with the teriflunomide/teriflunomide group during the open-label extension period is consistent with the known temporal risk profiles for these adverse events with teriflunomide. 23 Acute pancreatitis requiring permanent treatment discontinuation occurred in two participants in the TERIKIDS double-blind phase 15 and two participants in the open-label extension phase, which suggests the need for close observation. Teriflunomide was not associated with higher rates of pancreatitis in clinical trials involving adult participants, though post-marketing cases have been reported. 23 However, the serious nature of pancreatic adverse events requires prescribers to be properly informed of the risks of pancreatitis and apply appropriate vigilance when treating children with teriflunomide.
Study limitations were typical of open-label extension trials. Removal of treatment assignment blinding in the extension of the TERIKIDS study limits conclusions regarding efficacy findings. However, since MRI scans were assessed by a blinded rater, the risk of bias for MRI outcomes is probably low. It is also possible that a selection bias exists, since approximately 30% of participants discontinued the TERIKIDS study in the double-blind or open-label periods, mainly due to lack of efficacy or adverse events.
In conclusion, the long-term open-label extension of the TERIKIDS study supports the long-term beneficial radiological effects of teriflunomide in children with MS by reducing the risk of focal inflammatory activity, though further evidence would strengthen clinical efficacy findings. The safety and tolerability profile of teriflunomide was generally manageable over 192 weeks.
Supplemental Material
sj-docx-1-msj-10.1177_13524585241242050 – Supplemental material for Teriflunomide in pediatric patients with relapsing multiple sclerosis: Open-label extension of TERIKIDS
Supplemental material, sj-docx-1-msj-10.1177_13524585241242050 for Teriflunomide in pediatric patients with relapsing multiple sclerosis: Open-label extension of TERIKIDS by Tanuja Chitnis, Brenda Banwell, Ludwig Kappos, Douglas L Arnold, Kivilcim Gücüyener, Kumaran Deiva, Wenruo Hu, Stephane Saubadu, Myriam Benamor, Annaig Le-Halpere, Philippe Truffinet and Marc Tardieu in Multiple Sclerosis Journal
Footnotes
Acknowledgements
The authors thank the participants and their families for their involvement in, and commitment to, TERIKIDS and the clinical study team for the conduct of the study. Sanofi provided funding for medical writing and editorial support from Sally Laden, MS, and Conor F. Underwood, PhD, (Envision Pharma Group) based on input from the authors.
Data Availability Statement
Qualified researchers may request access to patient level data and related documents. Patient-level data will be anonymized, and study documents will be
redacted to protect the privacy of participants. Further details on Sanofi’s data-sharing criteria and process for requesting access can be found at: ![]()
Declaration of Conflicting Interests
The author(s) declared the following potential conflicts of interest with respect to the research, authorship, and/or publication of this article:
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This study was funded by Sanofi.
Supplemental Material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.