
Abstract
Background:
In previous studies, teriflunomide significantly reduced the annualised relapse rate (ARR) and disability progression.
Objective:
This phase 3, rater-blinded study (NCT00883337) compared teriflunomide with interferon-beta-1a (IFNβ-1a).
Methods:
Patients with relapsing multiple sclerosis were randomised (1:1:1) to oral teriflunomide 7-or 14mg, or subcutaneous IFNβ-1a 44µg. The primary composite endpoint was time to failure, defined as first occurrence of confirmed relapse or permanent treatment discontinuation for any cause. Secondary endpoints included ARR, Fatigue Impact Scale (FIS) and Treatment Satisfaction Questionnaire for Medication (TSQM). The study was completed 48 weeks after the last patient was randomised.
Results:
Some 324 patients were randomised (IFNβ-1a: 104; teriflunomide 7 mg: 109; teriflunomide 14 mg: 111). No difference in time to failure was observed. There was no difference in ARR between teriflunomide 14 mg and IFNβ-1a, but ARR was significantly higher with teriflunomide 7 mg. FIS scores indicated more frequent fatigue with IFNβ-1a, though differences were only significant with teriflunomide 7 mg. TSQM scores were significantly higher with teriflunomide. There were no unexpected safety findings.
Conclusion:
Effects on time to failure were comparable between teriflunomide and IFNβ-1a. There was no difference between teriflunomide 14 mg and IFNβ-1a on ARR, though ARR was higher with teriflunomide 7 mg. The teriflunomide safety profile was consistent with previous studies.
Keywords
Introduction
Teriflunomide is a once-daily, oral, disease-modifying therapy (DMT) for relapsing forms of multiple sclerosis (RMS). Teriflunomide selectively and reversibly inhibits dihydro-orotate dehydrogenase (DHODH), a mitochondrial enzyme in de novo pyrimidine synthesis required by rapidly dividing lymphocytes.1,2 Preclinical evidence indicates that teriflunomide exerts a cytostatic effect on stimulated lymphocytes in the periphery, reducing their availability to migrate into the central nervous system (CNS).3–6 As the pyrimidine demand of resting and slowly dividing cells (including memory lymphocytes) is met through the DHODH-independent salvage pathway, 7 teriflunomide may preserve protective immunity.8–10
In two pivotal phase 3 trials, the
In the TENERE (
Methods
Patients and procedures
This phase 3, multicentre, parallel-group, rater-blinded study enrolled patients 18 years of age and older who met McDonald criteria for MS, 13 had a relapsing clinical course with or without progression, and an Expanded Disability Status Scale (EDSS) score ≤5.5 at screening. 14 Patients had to be relapse free for 30 days prior to randomisation.
Exclusion criteria prohibited prior use of subcutaneous (SC) IFNβ-1a, teriflunomide, or leflunomide; prior or ongoing use of natalizumab, cladribine, mitoxantrone, or other immunosuppressants; or use of other interferons, glatiramer acetate, intravenous immunoglobulins, or cytokine therapy within 3 months. Patients were also excluded if they had other relevant systemic illnesses, were pregnant and/or breast-feeding, or planning to conceive.
The study was conducted in accordance with the 18th World Health Congress Recommendations, Declaration of Helsinki, and all applicable amendments. The protocol was approved by independent ethics committees and institutional review boards, and complied with local laws and regulations. All patients provided informed consent.
Patients were randomised 1:1:1 to teriflunomide 7 mg or 14 mg (double-blind) or IFNβ-1a (open-label), and stratified by country (Americas, Eastern Europe, Western Europe and Africa) and baseline EDSS score (≤3.5 or >3.5).
The treating neurologist was responsible for patient selection, medication administration, managing AEs, and relapse and safety assessments, while an examining neurologist scored the Functional Systems (FS) and EDSS. The examining neurologist remained blinded to treatment and associated AEs.
Teriflunomide 7 mg or 14 mg was administered as a single oral dose with or without food. Patients who discontinued teriflunomide underwent an accelerated elimination procedure using cholestyramine 8 g three times daily or activated charcoal powder 50 g four times daily for 11 days.
IFNβ-1a was administered as a SC injection three times per week, with the dose titrated from 8.8 µg for the first 2 weeks to 22 µg for the next 2 weeks, and 44 µg until study completion. When the 44 µg dose was not tolerated, the dose was reduced to 22 µg.
The study was completed 48 weeks after the last patient was randomised, resulting in a variable duration of follow-up.
Study evaluations
The primary composite endpoint was time to failure, defined as first occurrence of confirmed relapse or permanent treatment discontinuation for any cause. This endpoint was chosen to evaluate effectiveness, an endpoint relevant to real-world experience, as it accounts for factors related to efficacy, safety and tolerability. Relapse criteria required the appearance of a new clinical sign/symptom or clinical worsening of a previous sign/symptom (previously stable for at least 30 days) that persisted for at least 24 hours without fever. Each relapse was confirmed by the treating neurologist based on the objective assessment of the examining neurologist. A confirmed relapse required a 1-point increase in each of two FS, a 2-point increase in at least one FS (excluding bowel/bladder and cerebral) or an increase of ≥0.5 points in EDSS score from the previous stable assessment. Scores for FS and EDSS were assessed at randomisation, Weeks 12, 24 and 36, and then every 12 weeks until end of treatment.
Secondary endpoints were ARR (number of confirmed relapses during the treatment period per patient-year), changes in patient-reported fatigue (using the Fatigue Impact Scale (FIS), with higher scores indicating worsening fatigue (range: 0–160))15,16 and treatment satisfaction (using the Treatment Satisfaction Questionnaire for Medication (TSQM, version 1.4), with domains for Effectiveness, Side-Effects, Convenience and Global Satisfaction (range: extremely dissatisfied–extremely satisfied)). 17 Scores for FIS and TSQM were documented at baseline (FIS only) and Weeks 12, 24, 36 and 48, and every 24 weeks thereafter.
Safety and tolerability were assessed using AE reporting, vital signs and laboratory assessments. Adverse event reports were collected at randomisation, Weeks 2, 6, 12, 18, 24, 36 and every 12 weeks thereafter. Vital signs were documented at screening, randomisation and every 12 weeks thereafter; clinical laboratory results were assessed throughout the study. Adverse events and vital signs were also recorded during unscheduled relapse visits.
Any patient with an ALT increase >3× the upper limit of normal (ULN; confirmed by retest within 48 hours) was required to discontinue treatment and undergo further monitoring until levels normalised. Any occurrence of ALT >8× ULN or potential Hy’s Law (ALT >3× ULN and total bilirubin >2× ULN) was reported as a serious AE requiring discontinuation. Confirmed neutrophil counts <1000 cells/µl, with or without signs of infection, also necessitated treatment discontinuation, as did confirmed serum amylase or lipase values of >5× ULN, with or without clinical pancreatitis.
Statistical analyses
A sample size of 100 randomised patients per treatment arm provided 81% power to detect a difference between teriflunomide and IFNβ-1a on time to failure, at a significance level of α=0.025 (specified for multiplicity consideration). The study was not powered to detect differences in the individual components of the primary endpoint. Hazard rates of 0.4186 for teriflunomide and 0.7440 for IFNβ-1a were assumed, with recruitment duration of ~1.5 years and average follow-up of 1.75 years per patient.18–20 Given the sample size, average follow-up and assumed ARR of 0.4 in the IFNβ-1a arm, the study would detect ~36% relative reduction in ARR at a 0.05 significance level. Efficacy analyses were conducted on the intent-to-treat (ITT) population, which included all randomised patients. The safety analysis included all randomised patients exposed to study medication.
Time to failure was analysed using a log-rank test, with treatment group as the test variable, and region and baseline EDSS as stratum variables. All individual follow-up data were included until either confirmed relapse or permanent treatment discontinuation. If no events occurred, patients were considered free of treatment failure and data were censored at their last visit. The Kaplan–Meier method was used to estimate the rate of treatment failure at Weeks 24, 48 and 96. If medication was never received or incorrectly administered, it counted as a treatment failure.
The ARR was analysed using a Poisson regression model with robust error variance, including total confirmed relapses prior to discontinuation as the response variable, and treatment group, EDSS strata and region as covariates. Log-transformed standardised treatment duration (last dose date – randomisation date + 1)/365.25 was included as an offset variable to account for differences in exposure.
A mixed-effect model with repeated measures (MMRM) including factors for treatment, EDSS strata, region, visit, treatment-by-visit interaction, baseline value and baseline-by-visit interaction, was used to analyse change in total FIS score from baseline to Week 48. The MMRM was also used to analyse TSQM at Week 48, and included factors for treatment, EDSS strata, region, visit and treatment-by-visit interaction.
To estimate compliance, the number of doses the patient took was divided by the number of doses planned for the treatment period.
Results
Study disposition and population
Enrolment began on 16 April 2009 and the last patient completed treatment on 14 September 2011. The ITT population included 324 patients (IFNβ-1a n=104, teriflunomide 7 mg n=109; teriflunomide 14 mg n=111). Median exposure was 60.1 weeks in the IFNβ-1a group, 66.6 weeks in the teriflunomide 7 mg group, and 64.2 weeks in the teriflunomide 14 mg group. Maximum exposure was ~115 weeks. Mean compliance was high (IFNβ-1a: 97.6%; teriflunomide 7 mg: 99.2%; teriflunomide 14 mg: 98.4%). Overall, 22.4% of patients discontinued treatment, primarily due to AEs (Figure 1).
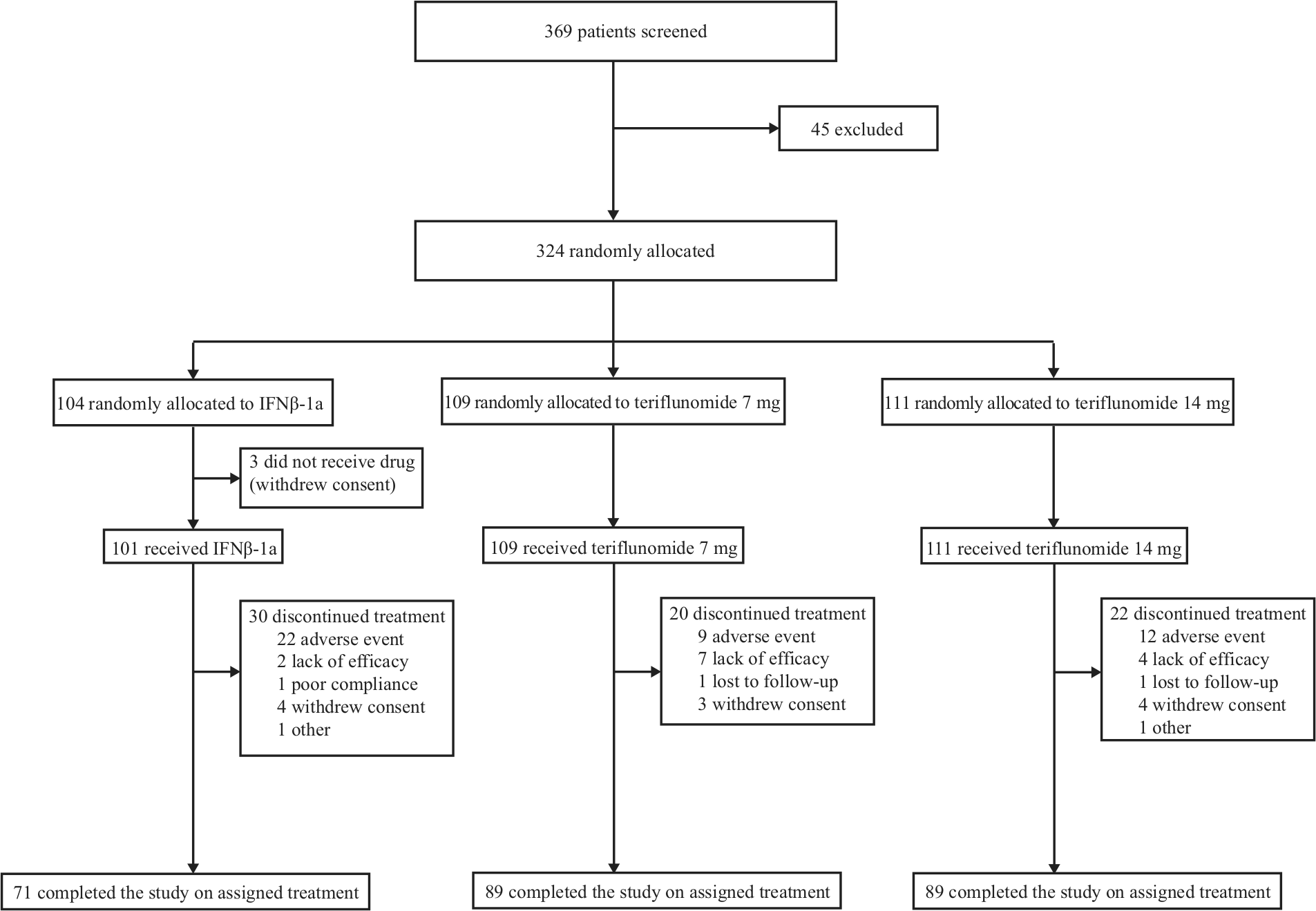
Patient disposition.
Baseline demographics and characteristics were balanced. However, there was significantly lower DMT use in the past 2 years in the teriflunomide 14 mg group compared with the IFNβ-1a group (Table 1).
Baseline demographics and disease characteristics.
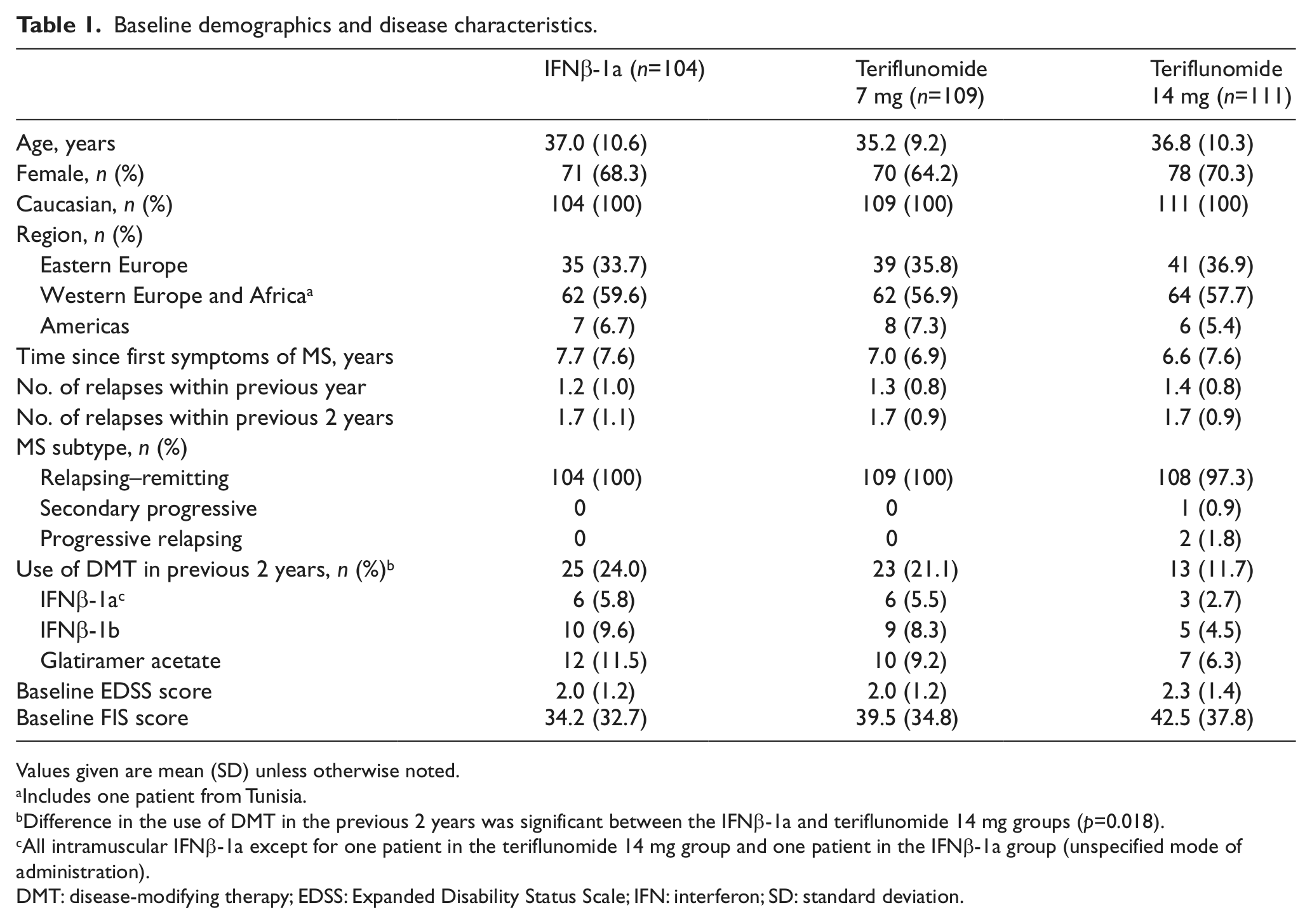
Values given are mean (SD) unless otherwise noted.
Includes one patient from Tunisia.
Difference in the use of DMT in the previous 2 years was significant between the IFNβ-1a and teriflunomide 14 mg groups (p=0.018).
All intramuscular IFNβ-1a except for one patient in the teriflunomide 14 mg group and one patient in the IFNβ-1a group (unspecified mode of administration).
DMT: disease-modifying therapy; EDSS: Expanded Disability Status Scale; IFN: interferon; SD: standard deviation.
Primary composite endpoint
No difference was found between either dose of teriflunomide and IFNβ-1a on time to failure. At Week 48, the cumulative percentage of estimated failures using the Kaplan–Meier method was 37% in the IFNβ-1a group, and 36% and 33% in the teriflunomide 7 mg and 14 mg groups (Figure 2). The contribution of permanent treatment discontinuation to the failure rate was highest in the IFNβ-1a group and lowest in the teriflunomide 7 mg group. In contrast, the fewest confirmed relapses were observed in the IFNβ-1a group (Table 2). Sensitivity analysis also showed results were consistent when all relapses (confirmed and not confirmed) were accounted for (Supplementary Materials), and when treatment effects were assessed across baseline strata.
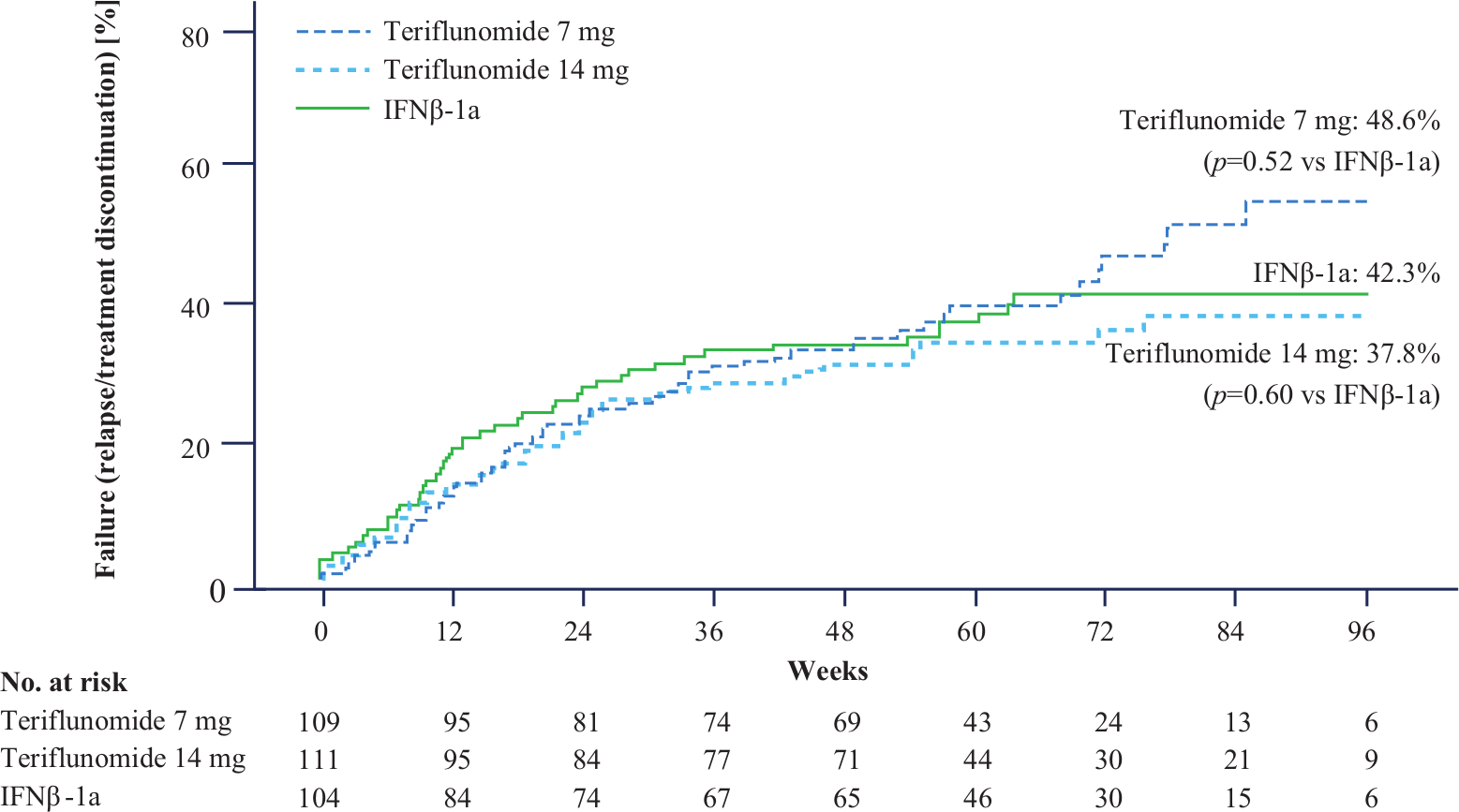
Time to failure.
Analysis of primary and secondary endpoints, ITT population.
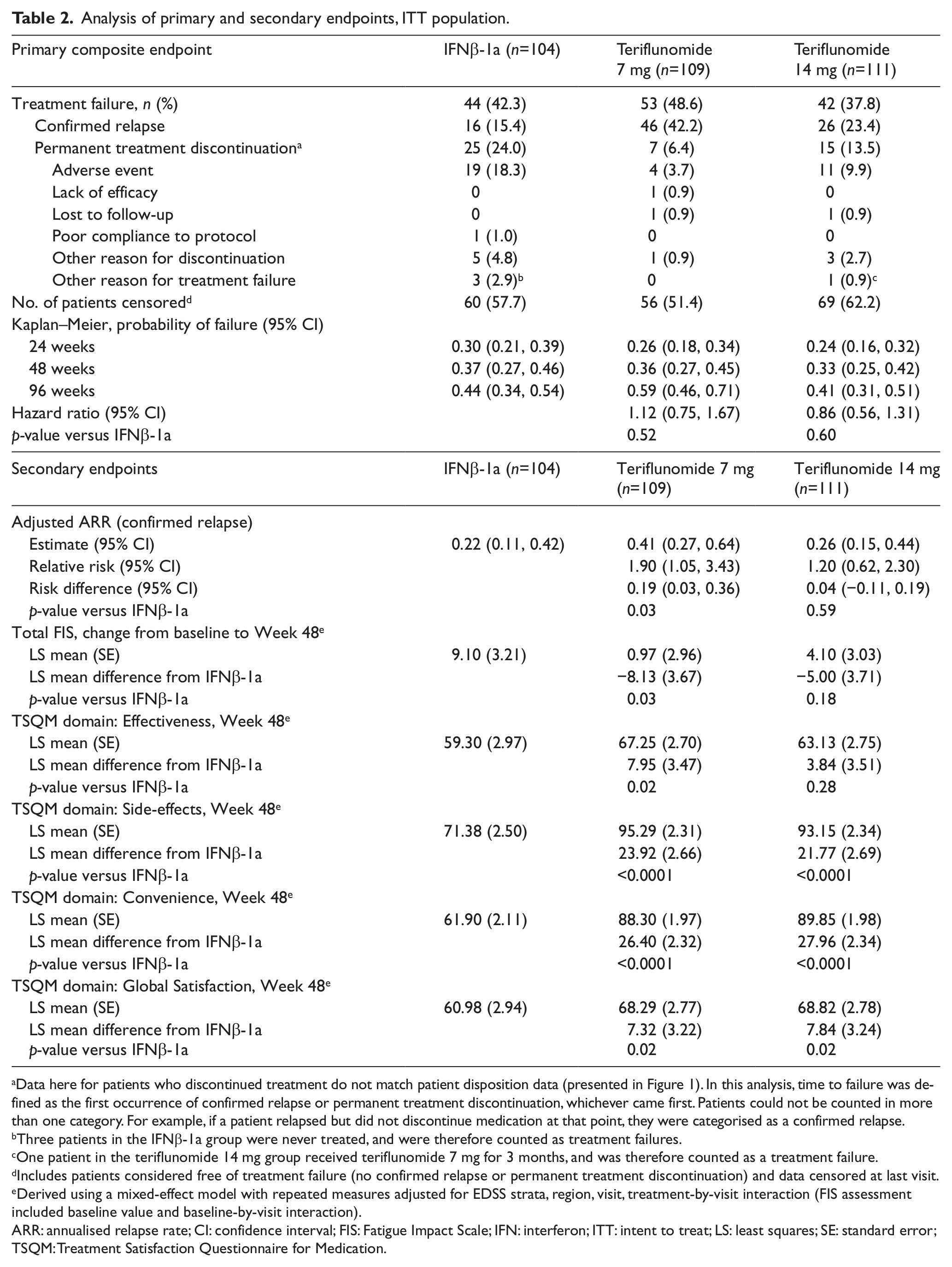
Data here for patients who discontinued treatment do not match patient disposition data (presented in Figure 1). In this analysis, time to failure was defined as the first occurrence of confirmed relapse or permanent treatment discontinuation, whichever came first. Patients could not be counted in more than one category. For example, if a patient relapsed but did not discontinue medication at that point, they were categorised as a confirmed relapse.
Three patients in the IFNβ-1a group were never treated, and were therefore counted as treatment failures.
One patient in the teriflunomide 14 mg group received teriflunomide 7 mg for 3 months, and was therefore counted as a treatment failure.
Includes patients considered free of treatment failure (no confirmed relapse or permanent treatment discontinuation) and data censored at last visit.
Derived using a mixed-effect model with repeated measures adjusted for EDSS strata, region, visit, treatment-by-visit interaction (FIS assessment included baseline value and baseline-by-visit interaction).
ARR: annualised relapse rate; CI: confidence interval; FIS: Fatigue Impact Scale; IFN: interferon; ITT: intent to treat; LS: least squares; SE: standard error; TSQM: Treatment Satisfaction Questionnaire for Medication.
Secondary endpoints
There was no difference in adjusted ARR between IFNβ-1a and teriflunomide 14 mg (0.22 versus 0.26, p=0.6), although ARR was significantly higher with teriflunomide 7 mg (0.41, p=0.03 versus IFNβ-1a) (Table 2). Nelson–Aalen estimates showed the occurrence of relapse was consistent over the treatment period, and sensitivity analysis including relapses occurring after treatment discontinuation was consistent with the primary analysis of relapse (Supplementary Materials).
The mean change from baseline to Week 48 in total FIS score indicated a greater adverse impact on fatigue with IFNβ-1a compared with either dose of teriflunomide. Differences from IFNβ-1a in total FIS score reached statistical significance only with teriflunomide 7 mg (Table 2).
Mean scores at Week 48 in the TSQM domains of Global Satisfaction, Side-Effects and Convenience were significantly improved with both doses of teriflunomide compared with IFNβ-1a. Improvements in Global Satisfaction primarily correlated with improved scores in the Side-Effects and Convenience domains associated with teriflunomide. Scores in the Effectiveness domain did not significantly differ between teriflunomide 14 mg and IFNβ-1a, but were lower with teriflunomide 7 mg (Table 2).
Safety and tolerability
The safety population included 321 patients (IFNβ-1a n=101; teriflunomide 7 mg n=110; teriflunomide 14 mg n=110). Overall occurrence of AEs was similar across groups (Table 3). Common AEs (≥10% in any group) reported more frequently with teriflunomide included nasopharyngitis, diarrhoea, hair thinning, paraesthesia and back pain. Influenza-like symptoms, ALT increases and headache occurred more frequently with IFNβ-1a (Table 4 and Supplementary Materials). We noted a similar incidence of serious AEs in the IFNβ-1a and teriflunomide 14 mg groups, and a higher incidence in the teriflunomide 7 mg group. With the exception of three cases of increased ALT in the teriflunomide 7 mg group, no serious AE was reported more than once. No deaths were reported during the study.
Adverse events.
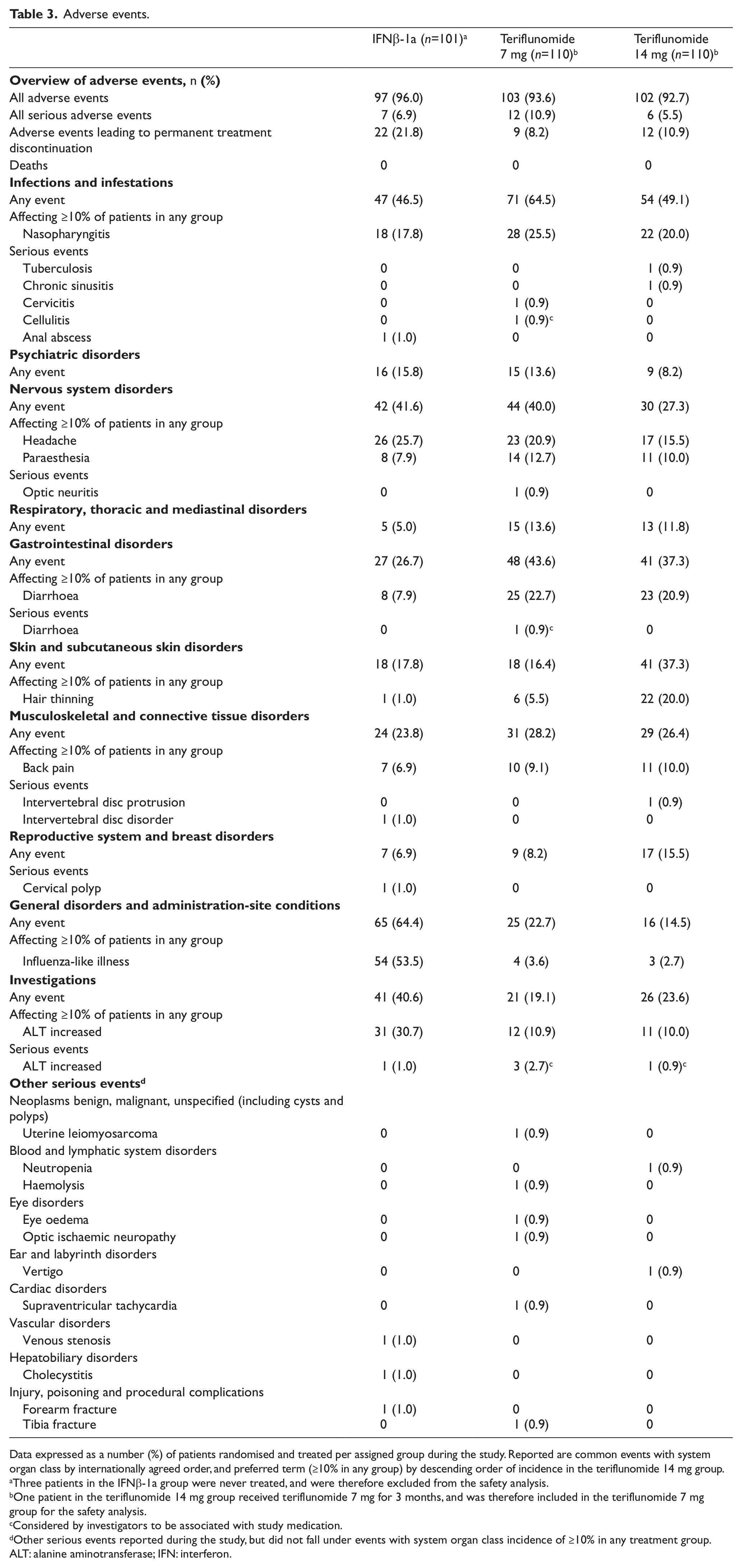
Data expressed as a number (%) of patients randomised and treated per assigned group during the study. Reported are common events with system organ class by internationally agreed order, and preferred term (≥10% in any group) by descending order of incidence in the teriflunomide 14 mg group.
Three patients in the IFNβ-1a group were never treated, and were therefore excluded from the safety analysis.
One patient in the teriflunomide 14 mg group received teriflunomide 7 mg for 3 months, and was therefore included in the teriflunomide 7 mg group for the safety analysis.
Considered by investigators to be associated with study medication.
Other serious events reported during the study, but did not fall under events with system organ class incidence of ≥10% in any treatment group.
ALT: alanine aminotransferase; IFN: interferon.
Overview of common adverse events (≥10% in any group).
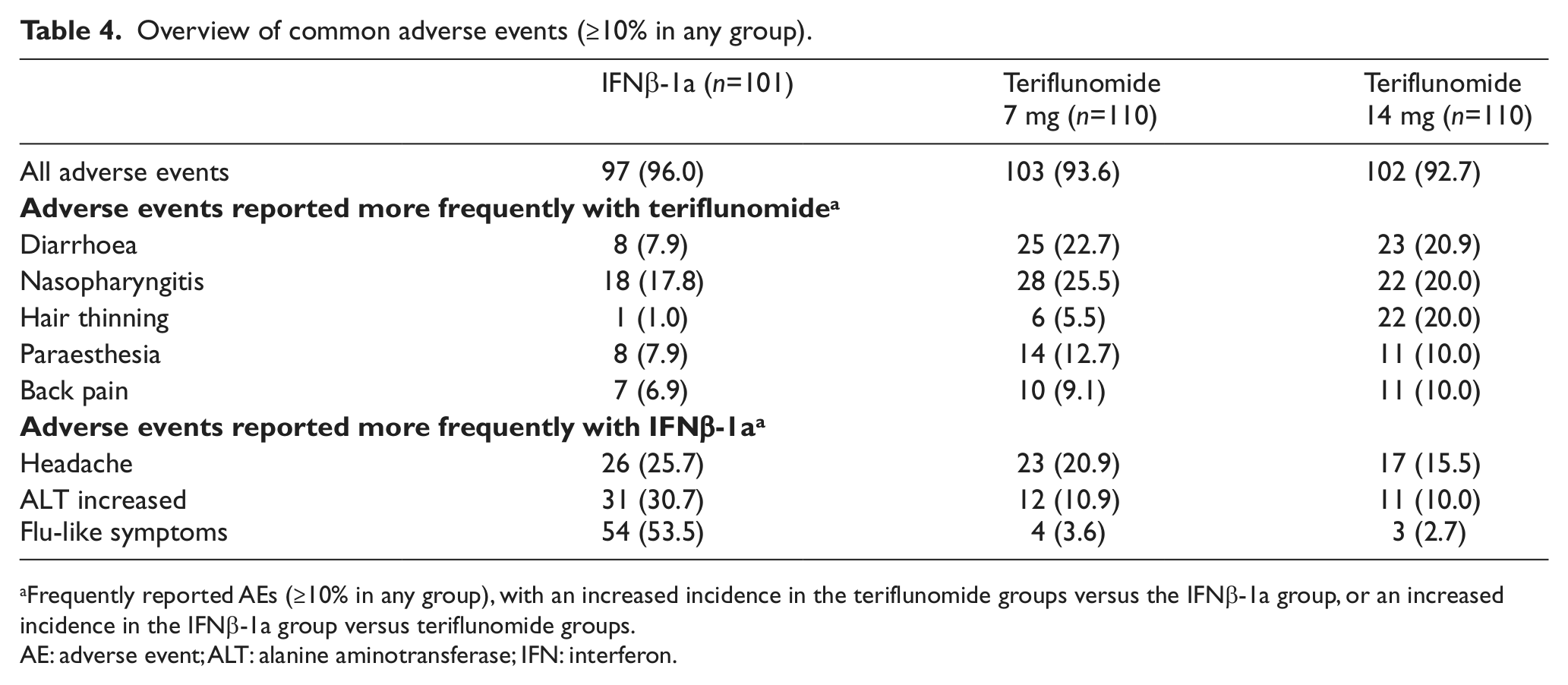
Frequently reported AEs (≥10% in any group), with an increased incidence in the teriflunomide groups versus the IFNβ-1a group, or an increased incidence in the IFNβ-1a group versus teriflunomide groups.
AE: adverse event; ALT: alanine aminotransferase; IFN: interferon.
Increased ALT was the most frequent cause of treatment discontinuation, and reported more frequently with IFNβ-1a than with teriflunomide (Tables 4 and 5). The majority of elevations in any group were ≤3× ULN, occurred within the first few months of treatment and generally normalised with continued treatment or following discontinuation (Table 6). All serious ALT elevations were asymptomatic and reversible, and no Hy’s law cases were observed.
Adverse events leading to permanent treatment discontinuation.
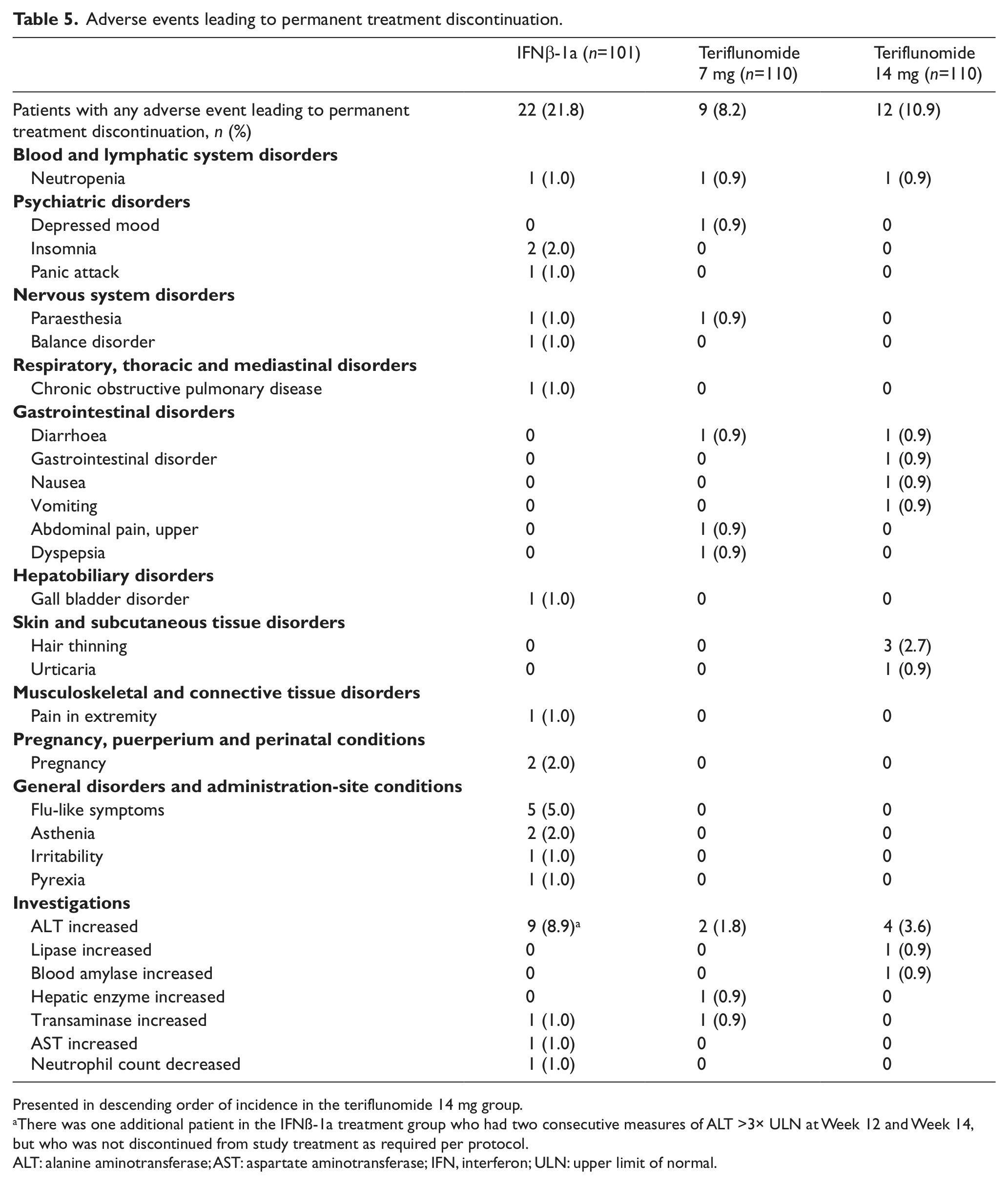
Presented in descending order of incidence in the teriflunomide 14 mg group.
There was one additional patient in the IFNß-1a treatment group who had two consecutive measures of ALT >3× ULN at Week 12 and Week 14, but who was not discontinued from study treatment as required per protocol.
ALT: alanine aminotransferase; AST: aspartate aminotransferase; IFN, interferon; ULN: upper limit of normal.
Laboratory evaluations.
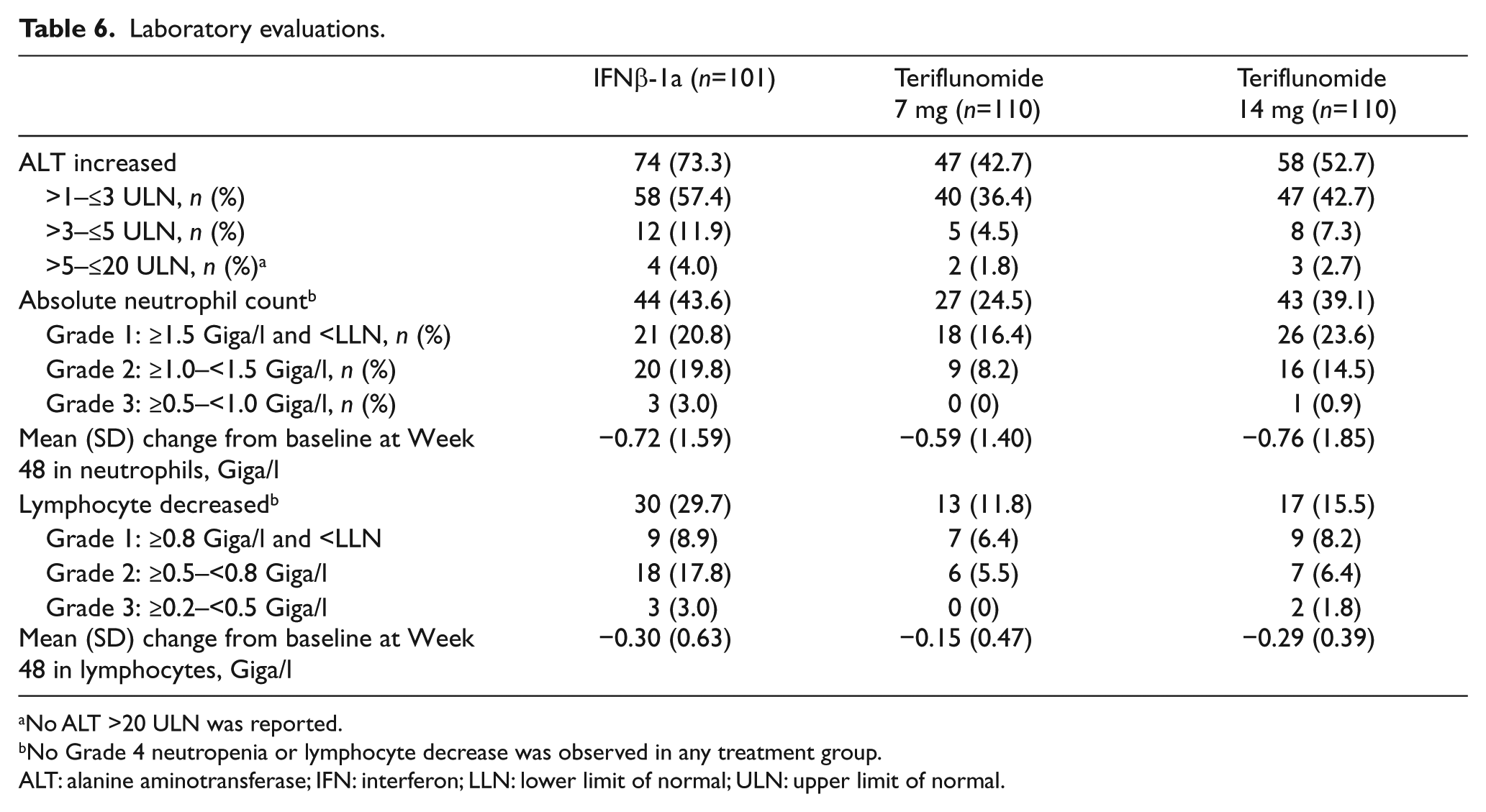
No ALT >20 ULN was reported.
No Grade 4 neutropenia or lymphocyte decrease was observed in any treatment group.
ALT: alanine aminotransferase; IFN: interferon; LLN: lower limit of normal; ULN: upper limit of normal.
White blood cell abnormalities were less frequent with teriflunomide 7 mg (4.5%) and 14 mg (5.5%) than with IFNβ-1a (10.9%). Mean decreases from baseline in lymphocytes were more pronounced with IFNβ-1a than with teriflunomide. Analysis showed decreases in neutrophil count from baseline occurred during the first 6 weeks in the teriflunomide and IFNβ-1a groups, and stabilised thereafter, with an average decrease of 0.7–0.8 Giga/l (Table 6 and Supplementary Materials).
There was a similar incidence of infections in the IFNβ-1a and teriflunomide 14 mg groups, and a higher incidence in the teriflunomide 7 mg group. Mild-to-moderate upper respiratory tract infections predominated, and there was no increased risk of serious infection with either treatment (1.0% in the IFNβ-1a group and 1.8% in each teriflunomide group).
Gastrointestinal events occurred more frequently with teriflunomide than with IFNβ-1a. The majority of cases were nausea and diarrhoea, which were rarely serious and infrequently led to treatment discontinuation. For the majority of patients with nausea and diarrhoea, full recovery was observed while continuing treatment. Most events were mild to moderate and did not require corrective therapy.
Hair thinning was more common with teriflunomide. The majority of cases occurred during the first 6 months and were mostly mild to moderate. Only three (2.7%) patients in the teriflunomide 14 mg group discontinued due to hair thinning (no patients in the teriflunomide 7 mg group). Most patients with hair thinning recovered while continuing treatment.
Influenza-like illness was reported more frequently with IFNβ-1a than with teriflunomide. No patients discontinued due to influenza in the teriflunomide groups, though 5.0% discontinued in the IFNβ-1a group.
There was a higher mean increase in systolic blood pressure from baseline to Week 48 with teriflunomide 14 mg (4.70 mmHg) and teriflunomide 7 mg (1.49 mmHg) compared with IFNβ-1a (0.04 mmHg), with a similar trend for diastolic blood pressure (14 mg: 4.39 mmHg; 7 mg: 0.99 mmHg; IFNβ-1a: 0.29 mmHg) (Supplementary Materials). No reports of hypertension were considered serious or resulted in treatment discontinuation.
Discussion
This trial did not demonstrate a significant difference between teriflunomide and IFNβ-1a on the primary composite endpoint of time to failure. Formal conclusions on effectiveness are challenging, as a larger patient population, longer treatment duration, and magnetic resonance imaging outcomes would be needed for a more robust comparison. However, the inclusion of relapse and permanent treatment discontinuation as components of the primary endpoint provides some clinical insight into the relative efficacy and tolerability of the two therapies.
Reductions in ARR were comparable in the teriflunomide 14 mg and IFNβ-1a groups, while ARR was significantly higher in the teriflunomide 7 mg group. The more robust effect observed with teriflunomide 14 mg compared with teriflunomide 7 mg is consistent with previous findings from phase 3 trials, in which a dose-response with teriflunomide was observed in ARR and significant decreases in disability progression were attained only with teriflunomide 14 mg.11,12
Despite the higher rate of treatment discontinuation with IFNβ-1a compared with teriflunomide, a major contribution of this imbalance on the ARR analysis was unlikely for several reasons. Firstly, variable treatment duration was accounted for in the statistical analysis. Secondly, the occurrence of relapses was consistent over time. Finally, a sensitivity analysis, which included relapses occurring after treatment discontinuation, was consistent with the main analysis of relapse.
An ad-hoc subgroup analysis evaluated the potential impact of previous MS medications on the primary composite endpoint and relapse. Outcomes did not detect a significant interaction between clinical responses and patients who had previously received interferon, and who may have experienced a suboptimal response. It may also be noted that while the examining neurologist was blinded to treatment, patients were un-blinded, which could have introduced a potential bias.
Adverse events leading to treatment discontinuation were highest in the IFNβ-1a group and were partly related to ALT increases. In a previous study of patients with MS receiving SC IFNβ-1a 44 µg three times weekly, ALT elevations were common. However, increases generally resolved spontaneously, or with a dose reduction or treatment interruption. 21 Our protocol did not allow for dosage modification, and any patient with confirmed ALT >3× ULN was required to permanently discontinue study treatment. The application of this rule across groups contributed to a higher rate of discontinuation in the IFNβ-1a group and it is unknown if, in the clinical setting, other approaches to managing liver enzyme elevations of this magnitude would have been effective. However, the low rate of ALT increases requiring discontinuation in the teriflunomide groups was consistent with previous clinical trial findings. 12
No unexpected safety concerns emerged, and the safety profile of teriflunomide was consistent with previous trials.12,19,22 Differences in safety and tolerability profiles are expected to factor in treatment selection, depending on patients’ individual characteristics and potential comorbidities. Teriflunomide and IFNβ-1a varied in tolerability, with flu-like symptoms more frequent with IFNβ-1a, and diarrhoea and hair thinning more common with teriflunomide. For these AEs common with teriflunomide, cases generally resolved with continued treatment, and discontinuation rates were low. Effects on laboratory evaluations, including liver enzymes and haematological parameters, were more pronounced with IFNβ-1a than with teriflunomide.
Non-adherence is common with DMTs and often relates to tolerability. In an observational study of patients with RMS (N=2566), 25% reported non-adherence or missing ≥1 DMT injection in the previous 4 weeks. Of these patients, 32% reported at least one injection-related reason. 23 Research indicates that oral medications and once-daily dosing regimens can improve adherence, and thereby treatment outcomes.24,25 In this study, teriflunomide-treated patients had greater treatment satisfaction, which may improve future adherence. 17 This prediction is in line with the expected benefit of an oral agent over an injectable therapy.
This study did not detect a difference in time to failure between either dose of teriflunomide and IFNβ-1a. A dose-response was observed with teriflunomide on ARR, as the 14 mg dose had a similar effect on relapse compared with IFNβ-1a, and the 7 mg dose was associated with a significantly higher relapse rate. Overall, patients reported greater satisfaction and less fatigue with teriflunomide than with IFNβ-1a. Based on these outcomes, teriflunomide can be considered as an alternative therapy for patients with RMS for whom treatment with interferon is being considered.
Footnotes
Acknowledgements
The Independent Data Monitoring Committee, Steering Committee, and Investigators for the
Conflict of interest
Patrick Vermersch has received consulting fees and/or research support from Bayer, Biogen Idec, GlaxoSmithKline, Merck Serono, Novartis, sanofi-aventis and Teva Pharmaceuticals.
Anna Czlonkowska has received research support and/or speaker support from Actelion, Bayer Health Care Pharmaceuticals, Bayer Schering Pharma, Biogen Idec, CSL Behring, Elan, GlaxoSmithKline, Lilly, Merck Serono, Novartis, Roche, sanofi-aventis, Teva, UCB and Wyeth.
Luigi ME Grimaldi has received consulting fees from Aventis, Biogen-Dompé AG, Merck Serono, Sanofi-Bayer Schering Pharma and Solvay Pharmaceuticals, Inc., and research support from Biogen-Dompé AG, Biogen Idec, Eisai Inc., Genzyme Corporation, Merck Serono, Ministero della Salute of Italy, Novartis, sanofi-aventis and Teva Pharmaceutical Industries Ltd.
Christian Confavreux has received consulting fees from Biogen Dompé, Biogen Idec, Gemacbio, Genzyme, Hertie Foundation, Novartis, sanofi-aventis, Teva and UCB; lecture fees from Bayer Schering, Biogen Idec, Merck Serono, Novartis, sanofi-aventis and Teva Pharma; research support from Bayer Schering, Biogen Idec, Merck Serono, Novartis, sanofi-aventis and Teva; and fees for membership of company Advisory Boards from Biogen Idec, Genzyme, Novartis, sanofi-aventis, Teva and UCB.
Giancarlo Comi has received consulting fees from Actelion, Bayer Schering, Merck Serono, Novartis, sanofi-aventis and Teva Pharmaceutical Ind. Ltd; and lecture fees from Bayer Schering, Biogen Dompè, Merck Serono, Novartis, sanofi-aventis, Serono Symposia International Foundation and Teva Pharmaceutical Ind. Ltd.
Ludwig Kappos has received research support from Actelion, Advancell, Allozyne, BaroFold, Bayer Health Care Pharmaceuticals, Bayer Schering Pharma, Bayhill, Biogen Idec, BioMarin, CSL Behring, Elan, Genmab, Genmark, GeNeuro SA, GlaxoSmithKline, Lilly, Merck Serono, MediciNova, Novartis, Novonordisk, Peptimmune, Roche, sanofi-aventis, Santhera, Teva, UCB and Wyeth, and from the Swiss MS Society, the Swiss National Research Foundation, the European Union, and the Gianni Rubatto, Novartis and Roche Research Foundations.
Tomas P Olsson has received consulting fees and/or research support from Biogen Idec, Merck Serono, and sanofi-aventis, and has participated in scientific advisory boards and/or speaking activities for Biogen Idec, Merck Serono and sanofi-aventis.
Myriam Benamor, Deborah Bauer and Philippe Truffinet are employees of Sanofi.
Meg Church is an employee of Fishawack Communications Ltd, which had been contracted to provide editorial support by Sanofi.
Aaron E Miller has received research support from Acorda Therapeutics, Biogen Idec, Genentech, Genzyme Corporation, Novartis, Osmotica, Roche, sanofi-aventis and Teva. He has received consulting fees from Acorda Therapeutics, Biogen Idec, EMD Serono, GlaxoSmithKline, Merck Serono, Novartis, Nuron Biotech, ONO and sanofi-aventis.
Jerry S Wolinsky has received consulting/speaker fees from Celgene, Consortium of MS Clinics, Genzyme, Hoffman LaRoche, Janssen RND, Medscape CME, Novartis, PRIME, sanofi-aventis, Serono Symposia International Foundation, Teva and Teva Neurosciences; royalties from Millipore (Chemicon International) Corporation; and research/contractual support from Genzyme, the National Institutes of Health, National MS Society and Sanofi.
Mark S Freedman has received research and educational grant support from Bayer Healthcare and Genzyme; honoraria/consultation fees from Bayer Healthcare, Actelion, Biogen Idec, EMD Canada, Novartis, Opexa Therapeutics, sanofi-aventis, Teva Canada Innovation and is a member of company Advisory Board/Board of Directors/or other similar group for Actelion, Bayer Healthcare, Biogen Idec, Celgene, Merck Serono, Novartis, Opexa Therapeutics, and sanofi-aventis.
Paul O’Connor has received consulting fees and/or research support from Actelion, Bayer, Biogen Idec, BioMS, Cognosci, Daiichi Sankyo, EMD Serono, Genentech, Genmab, Novartis, Roche, sanofi-aventis and Teva.
Funding
This study (NCT00883337) was funded by Genzyme, a Sanofi company. Editorial support was provided by Meg Church, Fishawack Communications, Ltd, also funded by Genzyme, a Sanofi company.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.