
Abstract
Central nervous system (CNS) demyelination is an uncommon observation in patients with Charcot-Marie-Tooth disease (CMT). Where it does occur, it is usually associated with X-linked CMT. We present a case of CMT type 1A with a likely de novo mutation who experienced initial symptoms, and subsequent exacerbation, of multiple sclerosis following respiratory infection. A review of the literature reveals that reports of CMT1A with CNS demyelination are rare. We propose that the mutations in the PMP22 gene result in an over-expression of PMP22 mRNA, which overcomes the normal suppression by miRNA species that occurs in the CNS. This abnormal expression of PMP22 protein may, in certain circumstances, exacerbate autoimmune responses to result eventually in CNS demyelination.
Introduction
Charcot-Marie-Tooth disease (CMT) is a peripheral neuropathy resulting in a progressive muscle weakness and reduced sensation in the distal limbs, hands and feet. It has well-established genetic etiology of which CMT type 1A accounts for approximately 50% of cases, most commonly involving duplication of the PMP22 gene encoding a peripherally-expressed myelin protein. 1 X-linked CMT (CMTX) is the second most common form, accounting for up to 15% of cases. Development of multiple sclerosis (MS) and other demyelinating disorders of the central nervous system are uncommon findings in people with CMT, most often observed in X-linked cases. 2 Such co-existing demyelinating disease is much rarer in association with CMT1A. Here we describe a case of MS in a male adolescent with CMT1A, review the literature relating to CNS disease in subjects with CMT1A and discuss the possible underlying mechanisms.
Case Report
A young male first presented at the age of 16 with an episode of sudden onset of numbness in the distal left hand and foot, without any impairment of limb muscle strength. The symptoms gradually worsened, peaking after about one week, and then resolved spontaneously. Two weeks prior to the onset, he had a respiratory tract infection. He had no prior medical history and did not smoke, drink alcohol, use illicit substances or have any indication of nutritional deficiency. Neurological examination revealed mild superficial sensory reduction in the left foot and absent deep tendon reflexes in the upper and lower limbs. The Expanded Disability Status Scale (EDSS) score was 1.0. MRI revealed several T2 flair hyperintense lesions affecting the left cervical spinal cord, brainstem, and the posterior limb of the right lateral ventricle, with no enhancement of the lesions (Figure 1A), and no significant abnormalities noted on susceptibility-weighted imaging (SWI) (Figure 2A). Contrast-enhanced lumbar MRI showed no enhancement of the nerve roots. Motor nerve conduction velocities were slowed, with prolonged distal latencies and reduced CMAP amplitudes; motor F-wave latencies were prolonged. Sensory nerve conduction velocities were likewise slowed, with reduced SNAP amplitudes or absent responses (Table 1). Cerebrospinal fluid cell count was 8x106/L, protein 1.119g/L, and negative results were obtained for autoimmune encephalitis antibodies in serum and cerebrospinal fluid, as well as for AQP4 antibodies, MOG antibodies, oligoclonal proteins and serum NF-155 antibodies. (A) 08/2020 MRI showed T2 flair sequences with multiple lesions involving both bilateral periventricular areas and the cervical spinal cord; (B) 04/2024 MRI revealed several T2 flair hyperintense lesions affecting the left cervical spinal cord, brainstem, and the posterior limb of the right lateral ventricle. more than 2020 (A) 08/2020 MRI SWI; (B) 04/2024 MRI SWI shows paramagnetic ring lesions around the lateral ventricles Nerve Conduction Studies Data Legend: MNCS Motor nerve conduction studies, SNCS Sensory nerve conduction studies, Lat Latency, Amp Amplitude, MCV Motor conduction velocity, Fmin Minimal latency of F wave,SCV Sensory conduction velocity, APB Abductor pollicis brevis, EDB Extensor digitorum brevis, Bl Bilateral, Ab Below, Pop popliteal, ADM Abductor digiti minimi, Erb Erb’s point, Dig Digit, EPL Extensor Pollicis Longus, ms Millisecond, uV Microvolt, m/s Meter per second.


Further genetic testing related to peripheral neuropathy identified a PMP22 locus duplication mutation with three repeats not found in the subject’s parents (Confirmed as biological parents of the patient during formal medical history inquiries conducted separately with each parent), who furthermore showed no related clinical manifestations. These results indicate a de novo mutation in the patient. After treatment with high-dose methylprednisolone, the patient experienced improvement and was discharged without long-term medication. Four years later, a week after the onset of an upper respiratory infection, the patient developed numbness in the left distal extremities, accompanied by mild weakness. The symptoms gradually worsened, peaking after about a week, and then resolved without intervention. The EDSS score was 1.5. A subsequent MRI showed T2 flair sequences with multiple lesions involving both bilateral periventricular areas and the cervical spinal cord, with location changes from four years previously (Figure 1B). Patchy changes were observed in the cervical spinal lesion (Figure 3), and SWI revealed paramagnetic ring lesions in the left lateral periventricular lesion (Figure 2B). MRI T2 shows an abnormal high signal intensity lesion in the left side of the spinal cord at the C2/C3 vertebral level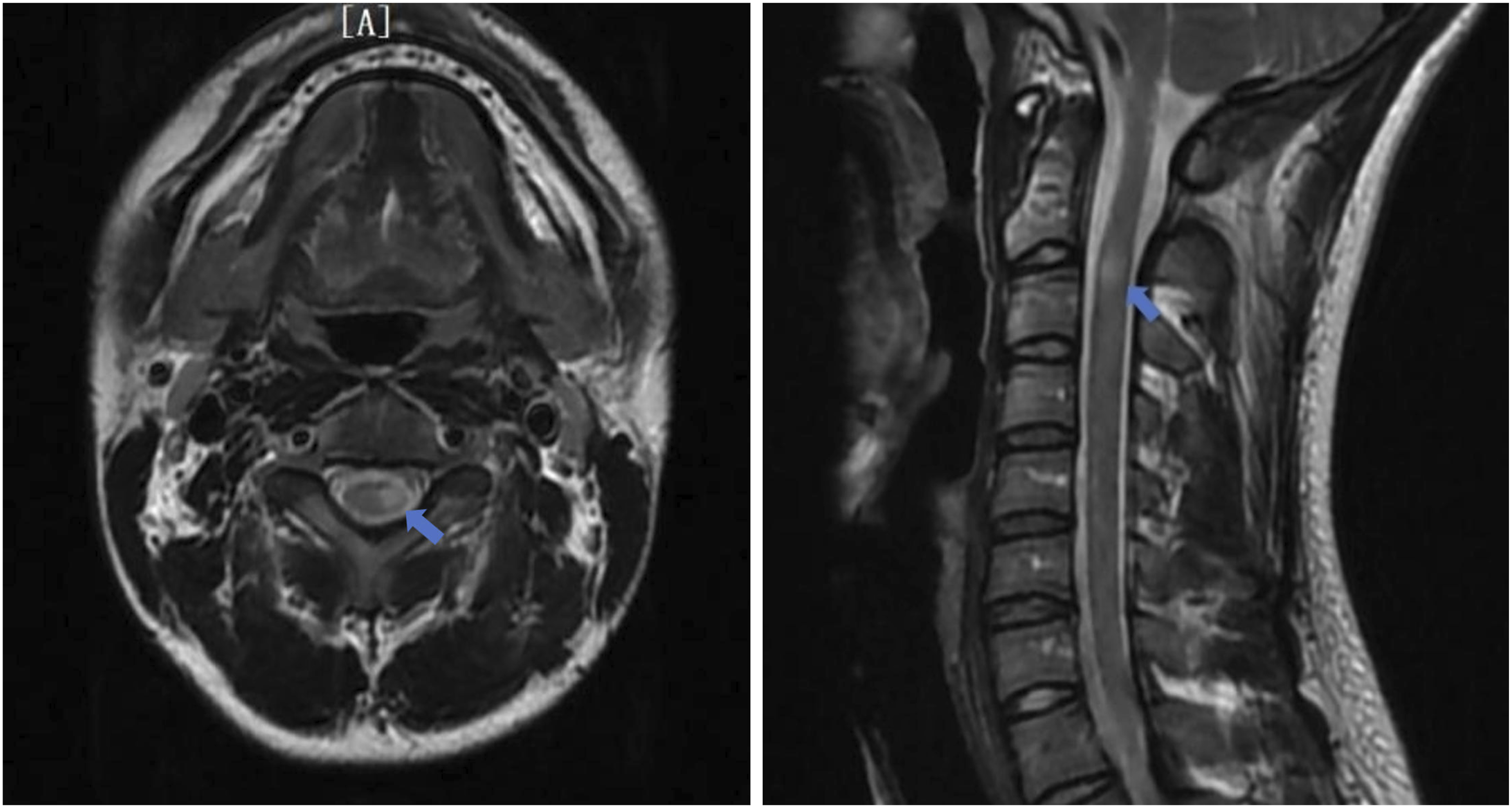
Re-examination of the cerebrospinal fluid showed that the AQP4 antibody, GFAP antibody, and MOG live cell antibody in serum and cerebrospinal fluid were all negative, and the oligoclonal protein was still negative. It was found that, in addition to localized sensory disturbances, the patient had reduced tendon reflexes in the limbs, slight high arches of the feet, and signs of muscle atrophy in the hands. Repeat electromyography showed no significant changes compared with 08/2020.
The patient was diagnosed with relapsing-remitting multiple sclerosis and CMT type 1A. After receiving further therapy with high-dose steroids, he underwent immunomodulation treatment with ofatumumab. After this treatment the patient’s symptoms fully resolved, with an EDSS score of 0.
Discussion
In this case, the initial episode, as well as the acute exacerbation four years later, occurred shortly after a respiratory infection. While a substantial proportion of MS exacerbations occur following an infection, 3 we did not identify any indication of systemic inflammation. Nevertheless an influence on immune response resulting in a disinhibition of autoimmune activity might be a consequence, perhaps in relation to sub-optimal vitamin D concentrations. 4 However the absence of any of the most likely autoantibodies is notable. During the second episode, multiple paraventricular white matter lesions and the iron ring-like appearance of the SWI sequence of the lesion were consistent with the “paramagnetic ring lesion” characteristic of multiple sclerosis, and the patient met the 2024 McDonald diagnostic standards despite being oligoclonal protein negative. Oligoclonal protein negativity has also been found in patients with other types of CMT combined with MS,2,5 and in one other case of CMT 1A. 6
The patient’s diagnosis of CMT-1A is supported by atrophy of the interosseous muscles of the hands and feet, loss of tendon reflexes in the limbs, frequent motor and sensory nerve demyelination changes in nerve conduction velocities, and a repeat mutation at PMP 22 by genetic testing. However, in the absence of anti-ganglioside and anti-PLP1 antibody testing we cannot completely rule out an atypical involvement of distinct inflammatory demyelinating disorder of the peripheral nervous system.
Multiple sclerosis is not a very rare disease, and its occurrence with CMT may be a coincidence. However, the presence of MS in various subtypes of CMT does suggest that the two diseases may be functionally associated. CMTX is more commonly associated with MS 2 ; here the gap junction connexin protein expressed by the mutant gene is found in both Schwann cells and CNS oligodendrocytes, 7 indicating how the dysfunction can be present in both peripheral and central nerves. CMTX with MS also shows abnormal features in the CNS, notably in the bilateral white matter of the posterior ventricles on MRI T2 sequence. 2
Reported Cases of CMT1A Combined With Central Nervous System Disorders

*This subject was positive for AQP4.
Comparing the features of the MS cases in Table 1, it is notable that all but one were positive for oligoclonal antibodies or, in the NMOSD case, for AQP4 antibodies. Other than that of Koros et al 6 our case is unique in not having detectable antibodies commonly associated with demyelinating disease. The other common feature of these two cases is the absence of a family history, although this may be coincidental; it is difficult to identify how de novo mutations may contribute to the unusual immunological profile.
While PMP22 is considered to be mainly expressed in the peripheral nervous system, PMP22 mRNA is found to be widespread in the human brain, particularly the corpus callosum. 15 However, protein expression is far more restricted, being detected primarily in spinal cord and not in the brain. This is thought to be due to the post-transcriptional effects of micro-interfering RNA, such as miR-9 or miR-29a, binding to a 3‘UTR sequence to suppress translation.16,17
Conclusion
In CMT1A, repeat mutations in the PMP22 gene may result in an over-expression of PMP22 RNA which then may not be fully suppressed by miRNA species. This is supported by the observation that a deletion in the 3’UTR sequence of the PMP22 gene can also result in a severe CMT phenotype. 17 A consequence would be the presence of PMP-22 in CMT1 oligodendrocytes – a feature of a genetic CMT animal model 18 – which has yet to be tested in human CMT1A. PMP22 proteins aberrantly produced in oligodendrocytes may undergo improper folding and retention. 19 The maturation and trafficking of CNS myelin membrane proteins such as Proteolipid protein-1 and Myelin oligodendrocyte glycoprotein require precise endoplasmic reticulum quality control, which depends on calnexin/calreticulin and endoplasmic reticulum-associated degradation resources. Retained misfolded PMP22 proteins may occupy these quality control resources, thereby impairing the maturation and trafficking of CNS myelin membrane proteins. 19 This dysfunction may then, in certain circumstances, exacerbate autoimmune response to result eventually in the degenerative process of MS.
Footnotes
Acknowledgements
We thank the participants for their contributions to this study.
Ethical Considerations
This single-patient case report uses fully de-identified information and does not constitute human subjects research; therefore, institutional review board approval was not required according to Huashan Hospital Fudan University policy.
Consent to Participate
Written informed consent to participate was provided by the patient.
Consent for Publication
Written informed consent for publication was obtained from the patient.
Author Contributions
Funding
Support for the investigations reported here was provided by the Shanghai Municipal Health Commission, award ID 202240383.
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Data Availability Statement
The data used in this study are available from the corresponding author upon reasonable request.