
Abstract
Background
Tetrahydrobiopterin (BH4) and its oxidized derivative dihydrobiopterin (BH2) were found to be strongly elevated in ME/CFS patients with orthostatic intolerance (ME + OI).
Objective
However, the molecular mechanism of biopterin biogenesis is poorly understood in ME + OI subjects. Here, we report that the activation of the non-oxidative pentose phosphate pathway (PPP) plays a critical role in the biogenesis of biopterins (BH4 and BH2) in ME + OI subjects.
Research Design and Results
Microarray-based gene screening followed by real-time PCR-based validation, ELISA assay, and finally enzyme kinetic studies of glucose-6-phosphate dehydrogenase (G6PDH), transaldolase (TALDO1), and transketolase (TK) enzymes revealed that the augmentation of anaerobic PPP is critical in the regulations of biopterins. To further investigate, we devised a novel cell culture strategy to induce non-oxidative PPP by treating human microglial cells with ribose-5-phosphate (R5P) under a hypoxic condition of 85%N2/10%CO2/5%O2 followed by the analysis of biopterin metabolism via ELISA, immunoblot, and dual immunocytochemical analyses. Moreover, the siRNA knocking down of the taldo1 gene strongly inhibited the bioavailability of phosphoribosyl pyrophosphate (PRPP), reduced the expressions of purine biosynthetic enzymes, attenuated GTP cyclohydrolase 1 (GTPCH1), and suppressed subsequent production of BH4 and its metabolic conversion to BH2 in R5P-treated and hypoxia-induced C20 human microglia cells. These results confirmed that the activation of non-oxidative PPP is indeed required for the upregulation of both BH4 and BH2 via the purine biosynthetic pathway. To test the functional role of ME + OI plasma-derived biopterins, exogenously added plasma samples of ME + OI plasma with high BH4 upregulated inducible nitric oxide synthase (iNOS) and nitric oxide (NO) in human microglial cells indicating that the non-oxidative PPP-induced-biopterins could stimulate inflammatory response in ME + OI patients.
Conclusion
Taken together, our current research highlights that the induction of non-oxidative PPP regulates the biogenesis of biopterins contributing to ME/CFS pathogenesis.
Plain Language Summary
Tetrahydrobiopterin (BH4) metabolism is tightly regulated in a healthy individual. Recently, our research showed that BH4 level is upregulated in the plasma samples of ME/CFS patients with orthostatic intolerance. While investigating the molecular mechanism, our current study identified that the pentose phosphate pathway (PPP) induction is critical for the upregulated expression of BH4. A novel hypoxia-based cell culture model is introduced to study PPP in human microglial cells. Subsequently, a comprehensive RNA array study, different immunoassay, and biochemical analyses of enzyme activities confirmed that the induction of non-oxidative PPP in microglial cells enhanced expressions of PPP-regulatory genes and enzymes, induced enzyme activities, activated purine biosynthesis, and finally upregulated biopterin biogenesis.
Introduction
Myalgic Encephalomyelitis/Chronic Fatigue Syndrome (ME/CFS) is a chronic multisystem illness characterized by severe muscle fatigue, pain, dizziness, and cognitive impairment.1,2 A subset of ME/CFS patients 3 exhibit a sudden and significant reduction of effective blood flow causing dizziness and fainting while maintaining an upright condition. 4 This condition is often termed as orthostatic intolerance (OI). Despite intense investigation, the molecular mechanism of OI is not known.
Tetrahydrobiopterin (BH4) is a cofactor that regulates the enzymic reactions of all hydroxylase enzymes5-7 with a wide range of metabolic implications including catabolic conversions of amino acids to neurotransmitters, and mitochondrial energy molecules.8-10 Moreover, BH4 is also a cofactor of nitric oxide synthase enzyme11,12 and is required for the endothelial synthesis of vasodilative soluble factor nitric oxide. Consequently, induction of BH4 biosynthesis stimulates the production of endothelial nitric oxide resulting in a strong vasodilative response, which often leads to the development of pulmonary hypotension. 13 In addition to that, BH4 is also a cofactor of the inducible nitric oxide synthase (iNOS) enzyme 14 that directly contributes to the inflammatory response in macrophages and microglia. A recent study 15 indicated that the uncontrolled upregulation of BH4 induces mitochondrial dysfunction, cognitive impairment, 16 immune dysregulation, 17 progression of inflammatory and autoimmune diseases, 18 and chronic pain. 18 Nevertheless, the metabolic conversion of BH4 to BH2 upregulates the synthesis of reactive nitrogen species,19,20 directly contributing to the inflammatory response in cells. 21 Recently, our study 22 has demonstrated that ME/CFS subjects with OI (ME + OI) are associated with the elevated expression of BH4 suggesting the potential role of upregulated BH4 in the pathogenesis of OI. However, the molecular mechanism of BH4 upregulation in ME + OI subjects is not known.
The pentose phosphate pathway (PPP) 23 is an alternative metabolic pathway of glucose metabolism 24 that utilizes six-carbon glucose-6-phosphate as a substrate to generate numerous 5-carbon (C5) intermediates such as fructose-6-phosphate, ribulose-5-phosphate, ribose-5-phosphate, and xylulose-5-phosphate. 25 Some of the metabolic intermediates of PPP such as fructose 6 phosphate and a 3-carbon (C3) glyceraldehyde-3-phosphate merge into glycolysis for energy production. 26 PPP operates in both oxidative and non-oxidative phases. At the first oxidative phase of PPP, glucose-6-phosphate converts into an oxidized lactone and also generates 1 molecule of reduced NADPH. After that, the oxidized lactone converts into C5 ribulose-5-phosphate and merges with glycolysis. NADPH increases reductive potential in the cell. These metabolites therefore contribute to the aerobic or oxidative fate of glucose molecules. However, the non-oxidative pathway of PPP 27 leads to the formation of ribose-5-phosphate, which is critical for the synthesis of purine intermediate GTP. Since GTP is also a primary precursor of BH4 biosynthesis, 28 we want to investigate if the augmentation of PPP upregulates BH4 biosynthesis in ME + OI subjects.
Our mRNA-based microarray study followed by the real-time PCR and quantitative ELISA analyses confirmed that the expressions of PPP enzymes were highly upregulated in ME + OI subjects. Subsequent enzyme kinetic studies of PPP enzymes such as glucose-6-phosphate dehydrogenase (G6PDH) and transketolase (TK) in plasma indicated that the non-oxidative PPP is highly operative in ME + OI subjects. Next, the implementation of non-oxidative PPP in C20 microglia stimulated the expression of GTPCH1, upregulated the synthesis of BH4, and induced the expression of iNOS. Interestingly, our siRNA-mediated silencing of the transaldolase (taldo1) gene followed by the evaluation of BH4 expression, iNOS activation, and nitric oxide production demonstrated that the augmentation of non-oxidative (non-ox) PPP followed by the upregulation of BH4 is critical in the pathogenesis of OI.
Results
Upregulation of BH4 Biosynthetic Genes in ME + OI Subjects
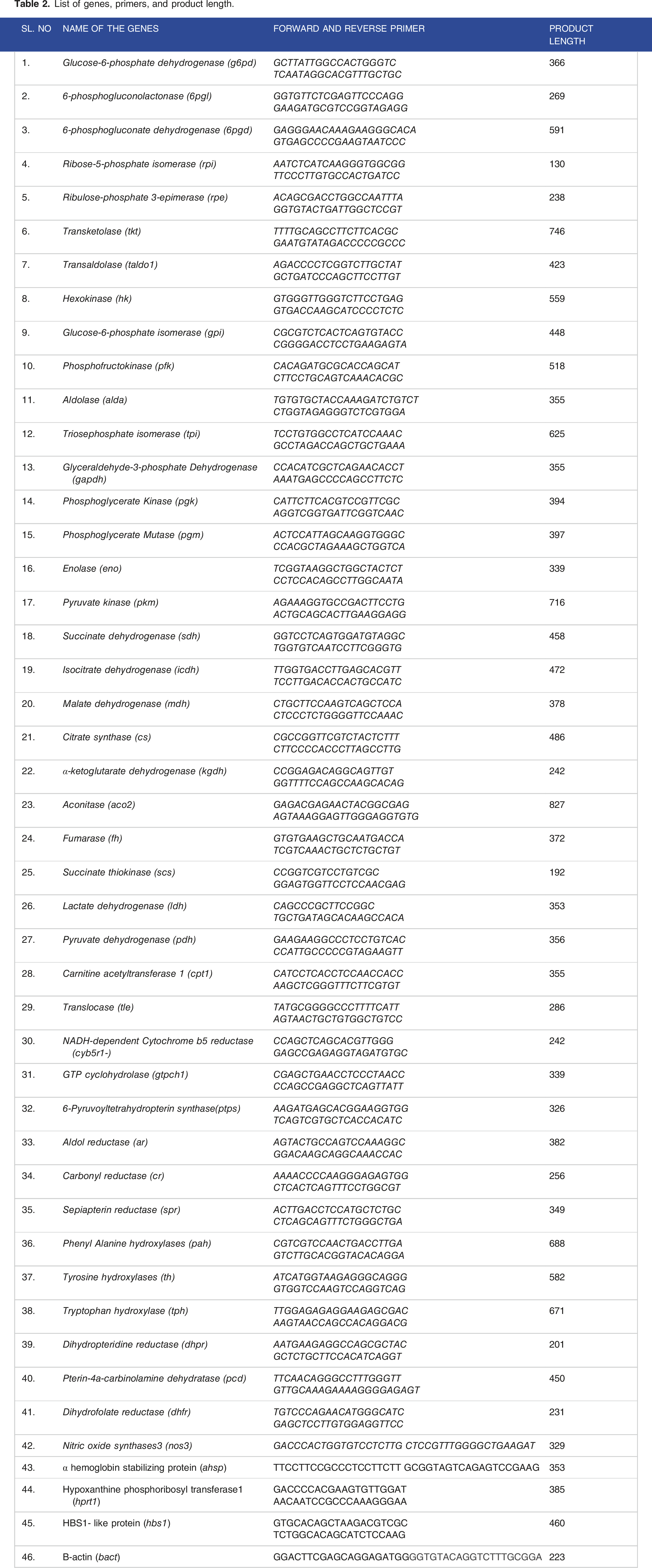
To understand the role of PPP in BH4 expression, we first performed a comprehensive microarray screening of 46 genes from the PAXgene total RNA samples of ME + OI (n = 4) subjects and age-matched healthy controls (HC) (Figure 1(A)). These genes directly or indirectly regulate BH4 expression. Thirty genes were selected from PPP-associated pathways of glucose metabolism. These genes are g6pd, 6pgl, 6pgd, rpi, rpe, tkt, taldo, hk, gpi, pfk, alda, tpi, gapdh, pgk, pgm, eno, pkm, sdh, icdh, mdh, cs, kgdh, aco2, fh, scs, ldh, pdh, cpt1, tle and cyb5r1. Twelve genes were selected from direct, regenerative, and salvage biosynthetic pathways of BH4. These genes are gtpch1, ptps, ar, cr, spr, pah, th, tph, dhpr, pcd, dhfr and nos3. Four genes were selected as housekeeping genes including ahsp, hprt1, hbs1, and bactin1 Gene array strategy to monitor BH4-regulating genes in RNA samples of healthy controls (HC) and ME/CFS subjects with orthostatic intolerance (ME + OI). (A) Blood samples were collected in a PAXgene Tube, processed for mRNA, and utilized for mRNA-based gene array study as described under the method section. An mRNA-based gene array was performed with 42 glucose metabolic genes and 4 housekeeping genes in ME + OI and age-/gender-matched healthy controls (n = 3 out of 4). Normalized Ct values were displayed in a representative heatmap (drawn by Morpheus software in an online server of Broad Institute) followed by clustering analysis. The color gradient was selected from green to red indicating low to high expression of genes. A cluster of blue arrows on the dendrogram indicated the upregulated genes in ME + OI subjects (n = 3). These genes encode for 6GPD or G6PDH (glucose 6 phosphate dehydrogenase), DHPR (dihydropteridine reductase), lactate dehydrogenase (LDH), CR (carbonyl reductase), phenylalanine hydroxylase (PAH), GTPCH1 (GTP cyclohydrolase1), DHFR (dihydrofolate reductase), NOS3 (nitric oxide synthase1or eNOS). (B) Expressions of several housekeeping genes such as ahsp (encodes for α hemoglobin stabilizing protein or AHSP), hprt1 (encodes for Hypoxanthine phosphoribosyl transferase 1or HPRT1), hbs1(encodes for HBS1-like protein), and bactin (encodes for β-actin) were monitored by real-time PCR analysis in both HC and ME + OI samples to nullify the loading error and confirm the quality of mRNA samples. Results are mean ± SEM of 3 different experiments. Ns = not significant. (C) The Venn diagram summarizes the numbers of downregulated (green circle; 5 genes), upregulated (red circle; 20 genes), and unaltered (shared circle; 17 genes) genes. Expression of the tryptophan hydroxylase (tph) gene was not included by the software due to its detection being much lower than the cut-off value. Volcano plot analyses represent the realtime mRNA validations of (D) G6PDH (*P < 0.05 by unpaired t-test), (E) TALDO1(**P < .01 by unpaired t-test), (F) 6PGL or 6-phosphogluconolactonase (no significance), (G) LDH (**P < .01 by unpaired t-test), (H) HK or hexokinase(***P < .001 by unpaired t-test), (I) GAPDH or glyceraldehyde-3-phosphate dehydrogenase (*P < 0.05 by unpaired t-test), (J) GTPCH1 (*P < 0.05 by unpaired t-test), in ME + OI and age-/gender-matched HC (n = 10 per group). (K) Semi-quantitative RT-PCR amplifications followed by resolving PCR products with 1.5% agarose gel and imaging in SmartDoc imaging system (Accuris Instruments) using smart glow dye demonstrated mRNA expressions of genes encoding G6PDH and GTPCH1 in HC and ME + OI subjects. The expression β-actin was visualized as a housekeeping gene. Results are confirmed after 3 different experiments. Activation of G6PDH, a rate-limiting enzyme of PPP, in ME + OI subjects. (A) The enzymic activity of G6PDH (n = 9 HC/n = 10 ME + OI) and (B) NADP/NADPH (n = 10 HC/n = 10 ME + OI) levels were monitored in the PBMC lysates of ME + OI and HC subjects. An Unpaired t-test was performed between the 2 groups. The resultant significance of the mean demonstrated **P < .001 and *P < .05 vs HC groups.

Upregulation of Non-Oxidative PPP in ME + OI Subjects
Our gene array and subsequent real-time PCR studies indicated that ME + OI subjects are associated with induced non-oxidative PPP. To confirm the upregulation of non-oxidative PPP, we performed ELISA analyses of 2 non-oxidative enzymes of PPP including transaldolase (TALDO) (Figure 3(A)) and transketolase (TK) (Figure 3(B)). Accordingly, expressions of TALDO, but not TK, were found to be significantly upregulated in ME + OI cases compared to healthy controls suggesting that non-oxidative PPP could be highly operative in ME + OI cases (n = 10). Although TK expression was not altered, the enzymic activity assay of TK displayed a significantly higher activation in ME + OI subjects (n = 8 per group) compared to controls (Figure 3(C)) suggesting that the activation of non-oxidative PPP could be critical in the pathogenesis of ME + OI. Augmentation of anaerobic PPP in ME + OI subjects and the introduction of a novel cell culture strategy to study anaerobic PPP. ELISA analyses of (A) TALDO (*P < 0.05 by an unpaired t-test) and (B) TK (ns = no significance) were performed in PBMCs of HC (n = 9) and ME + OI subjects (n = 10) as described in the method section. (C) TK enzyme activity was measured in a kinetic mode at a 30-second interval for 45 minutes in lysed PBMCs of HCs (n = 8) and ME + OI subjects (n = 8). An unpaired t-test was adopted to test the significance of the mean between 2 groups (****P < .0001). (D) A schematic diagram of a cell culture strategy to induce anaerobic PPP in cells. Evaluations of (E) TALDO1 and (F) TK expressions in C20 microglial cells treated with 2, 5, and 10 μM R5P under regular and hypoxic conditions. A two-way ANOVA was adopted to test the significance of means between control and R5P-treated groups. The independent variables or effectors are treatment (control vs R5P) and condition (regular vs hypoxia). #P < 0.05, *P < 0.05 vs regular control and hypoxia control respectively. TK enzyme activities were measured in C20 microglial cells treated with R5P under (G) regular and (H) hypoxic conditions for 24 hrs. An unpaired t-test was adopted to compare the significance of the mean between groups, which resulted in ***P < .001 vs regular control and ****P < .0001 vs hypoxic control. Results are mean ± SD of 3 independent experiments. Immunofluorescence analyses of TALDO1 (green) and β-actin (red) in C20 microglial cells treated with increasing doses of R5P under (I) regular and (J) hypoxic conditions. Nuclei were stained with DAPI.
To study the role of non-oxidative PPP in the pathogenesis of ME + OI, we devised a novel cell culture model (Figure 3(D)), in which cells will be treated with Ribose-5-phosphate (R5P) in a hypoxia chamber connected with mixed gas tank filled with 5%O2/10% CO2/85%N2. Induction of hypoxia was expected to induce the reductive potential in the cell, whereas ribose-5-phosphate (R5P) is the primary substrate of non-oxidative PPP. Treating cells with R5P in a hypoxia chamber for 24 hours ensures the non-oxidative induction of PPP. Microglial cells, brain-specific macrophages, were selected in this study because based on the human protein atlas database, macrophage profusely expresses TALDO1, the critical enzyme of non-oxidative PPP. Accordingly, we observed that R5P dose-dependently upregulated the expression of TALDO1 expression in C20 microglial cells kept under hypoxic, but not regular conditions (Figure 3(E)). Although increasing doses of R5P did not upregulate the expression of TK in hypoxic condition (Figure 3(F)) compared to regular condition, we observed a significant increase in TK enzyme activity in 10 μM R5P-treated microglia in hypoxic (Figure 3(H)), but not in regular condition (Figure 3(G)). Furthermore, our immunocytochemical study revealed that TALDO1 was significantly upregulated in human microglial cells upon stimulation with increasing doses of R5P for 24 hrs in a hypoxia chamber (Figure 3(J)), but not in a regular condition (Figure3(I)). The effect was observed maximum at 10 µM concentration.
TALDO and TK are reversible enzymes catalyzing both the turnover and synthesis of R5P. We hypothesized that upregulated activations of TALDO and TK via implementation of a reductive environment shifted the reaction equilibrium towards the augmented synthesis of R5P, a critical precursor of BH4 biosynthesis. In addition to the upregulated endogenous biosynthesis of R5P, the exogenous addition of R5P was expected to facilitate the downstream pathways required for the biosynthesis of BH4. For BH4 biosynthesis, R5P undergoes a series of enzymic reactions to produce guanine nucleotide (Figure 4(A)). R5P is first converted to phosphoribosyl pyrophosphate (PRPP) by the enzymic action of PRPP synthase (PRPPS). After that, PRPP is converted to inosine monophosphate (IMP), which in turn is converted to guanosine monophosphate (GMP) by the successive enzymic actions of IMP dehydrogenase (IMPDH) and GMP synthase (GMPS). First, we quantified the level of PRPP in the cell lysate of C20 microglia treated with 5 and 10 μM of R5P under both regular and hypoxic conditions. Previous reports29,30 suggest that the enzymic activity of PRPP synthase is highly elevated in myeloid cells under hypoxic conditions. Accordingly, we observed the PRPP level was highly elevated in the cell lysates of C20 microglial cells treated with 5 and 10 μM R5P under hypoxic, but not regular conditions (Figure 4(B)). Next, we performed a dual IF analysis of beta-actin (green) and IMPDH2 (red), which demonstrated that under regular condition, R5P treatment did not alter the expression of IMPDH2 (Figure 4(C); second panel) compared to regular control (Figure 4(C); First panel). However, the expression of IMPDH2 was found to be strongly elevated when treated with 10 μM of R5P (Figure 4(D); bottom panel) under hypoxic conditions. Finally, a dual IF analysis of beta-actin (green) and GMPS (red) (Figure 4(F)) displayed that C20 microglia were associated with a strong elevation of GMPS level under hypoxic conditions, but not under regular conditions (Figure 4(E)). Taken together, our results suggest that the supplementation of R5P under a less-oxygenated environment induced non-oxidative PPP, which in turn modulates the purine biosynthesis in microglial cells. Augmentation of non-aerobic PPP critically regulates the metabolic fate of Ribose-5-phospahte towards the synthesis of purine metabolites. 
Non-Oxidative PPP Regulates BH4 Biosynthesis
So far our results demonstrated that the implementation of non-aerobic PPP followed by the enzymic activations of TALDO and TK are critical to guide the metabolism of R5P through the purine biosynthetic route. Does this metabolic pathway play a critical role in the biogenesis of BH4? At first, we need to understand the regulation of BH4 biogenesis in ME/CFS. We performed a comprehensive ELISA analysis to explore these BH4-controlling factors in plasma samples of ME + OI subjects. First, we reanalyzed BH4 expression in n = 10 HC and n = 10 ME + OI plasma samples, normalized with respective protein concentration, and converted to pmol/mg unit after readjusting the quantitative data with the molecular weight of BH4. As we have shown before,
22
there was a strong upregulation of BH4 in the plasma samples of ME + OI subjects (Figure 5(A))compared to age-/gender-matched healthy subjects (HC). Next, we analyzed the protein expression of GTPCH1, a rate-limiting enzyme of de novo BH4 biosynthetic pathway. Similar to our microarray and PCR analyses, our ELISA study further corroborated that the protein expression of GTPCH1 was indeed strongly elevated in PBMCs of ME + OI subjects (Figure 5(B)). Dihydropteridine reductase (DHPR) enzyme converts quinonoid adduct of dihydrobiopterin (qBH2) to BH4 in the regenerative pathway. Accordingly, our ELISA analyses revealed that both BH2 (Figure 5(C)) and DHPR (Figure 5(D)) were strongly upregulated in the plasma and PBMCs of ME + OI subjects (n = 10 per group) respectively. The upregulation of DHPR and BH2 indicated that ME + OI pathogenesis may be associated with the altered metabolism of BH4 along with its upregulation. Anaerobic PPP upregulated the biosynthesis of BH4. (A) BH4 was quantified in the plasma samples of ME + OI (n = 10) and age-/gender-matched HC subjects. The BH4 levels in plasma were normalized with total protein concentration and further adjusted with the molecular weight of BH4 (241.25 g/mole). The final result was expressed by the pmol/mg unit. 
Interestingly, our absolute quantification analyses by ELISA revealed that even though both BH4 (Figure 5(A)) and BH2 (Figure 5(C)) levels are higher in ME + OI subjects, the BH2 level was found to be much higher than that of BH4. Therefore, along with BH4 upregulation, cellular homeostasis of BH4 was also disturbed in ME + OI subjects suggesting a possibility of a cellular nitrosylative stress 31 in ME + OI subjects.
To explore the regulations of these BH4-controlling cellular factors under induced non-oxidative PPP conditions, we performed protein expression analyses of GTPCH1(Figure 5(E) and (F)), BH4 (Figure 5(G)), and BH2 (Figure 5(H)) in increasing doses of R5P-treated C20 microglial cells kept in a hypoxia chamber for 24 hrs. Interestingly, Induction of non-oxidative PPP in C20 microglia upregulated expressions of GTPCH1, BH4, and BH2. On the contrary, no change of GTPCH1, BH4, and BH2 were observed when cells were incubated with R5P in non-hypoxic regular conditions. Similar to our plasma analyses, BH2 levels in that condition were found almost 1.5 times higher than BH4 suggesting the dysregulated nitric oxide synthesis in microglial cells. Taken together, our results suggest that the augmentation of non-oxidative PPP could activate the BH4 biosynthetic pathway in microglial cells.
To establish the direct role of non-oxidative PPP in purine biosynthesis followed by BH4 expression, we adopted a siRNA-mediated silencing strategy to knock down the taldo1 gene and then analyzed the expression of different molecular factors in purine and BH4 biosynthetic pathways in C20 microglia under induced hypoxic conditions. Briefly, the endogenous expression of TALDO1 in microglial cells was attenuated by transfection of taldo1 siRNA for 24 hrs followed by the treatment with 25 µM R5P in the hypoxia chamber for another 24 hrs. First, our immunoblot analysis of TALDO1 followed by the normalization with hypoxia-inducible factor-1 alpha (HIF-1α) and β-actin proteins validated that the siRNA efficiently knocked down the expression of TALDO1 (Figure 6(A)) without inducing cellular death (for full blot see Supplementary Figure 1). First, we observed that taldo1 gene knockdown significantly attenuated the R5P-induced expression of PRPP (Figure 6(B)). Second, IF analyses followed by quantification analyses revealed that the expressions of both IMPDH2 (Figure 6(C) and 6(D)) and GMPS (Figure 6(E) and (6F)) were strongly suppressed in R5P-treated C20 microglia once taldo1 gene is knocked out under hypoxic condition. Collectively, these results suggest that the augmentation of the non-oxidative phase of PPP directly facilitates the utilization of R5P in the purine biosynthetic pathway. Knocking down of anaerobic PPP by taldo1 siRNA ameliorated the expression of BH4 via inhibition of direct biosynthetic pathway. C20 microglial cells were treated with 0.25 μg of taldo1 siRNA (validated and inventoried by Thermofisher) for 24 hrs followed by the treatment with 10 μM R5P and immediate placement under hypoxic conditions. (A) After 24 hrs, cells were immunoblotted with TALDO1 antibody to see the taldo1 knock-down efficiency. The result was normalized with the expression β-actin. (B) Quantification of PRPP was performed in whole cell lysate of C20 microglial after treatment with 5 and 10 μM R5P under regular (green) and hypoxic conditions as discussed in the method section. *P < .05, **P < .001, and ***P < .0001 vs control. The significance of means between groups was analyzed by two-way ANOVA. (C) Dual IF analysis of β-actin (green) and IMPDH2 (red) in C20 microglia under hypoxic control, 10 μM R5P, and 10 μM R5P + taldo1 siRNA treatments. (D) Quantification analyses of mean fluorescence intensities (MFI) for IMPDH2 and corresponding β-actin were performed on 20 randomly selected cells, normalized, and then plotted as a histogram. One-way ANOVA was performed to test the significance of the mean between groups (***P < .0001). (E) Dual IF analyses of β-actin and GMPS in C20 microglia under similar treatment conditions. Nuclei were stained with DAPI. (F) Quantification studies followed by statistical analyses were performed as described under 6D. (G
Interestingly, silencing of the taldo1 gene strongly attenuated BH4 (Figure 6(G)) and GTPCH1(Figure 6(H)) expressions in C20 microglial cells once treated with 10 μM of R5P under hypoxic conditions.
To further confirm the direct role of nonaerobic PPP in the regulation of BH4, GTPCH1 expression was investigated by immunofluorescence study along with TALDO1 (Figure 6(I)). Interestingly, the expression of GTPCH1 was strongly inhibited in taldo1 siRNA-, but not control siRNA-transfected, R5P-treated microglial cells suggesting that the activation of non-oxidative PPP truly regulates BH4 biosynthesis via upregulation of GTPCH1.
Plasma-Derived BH4 Stimulates the Activation of Inducible Nitric Oxide Synthase and Nitric Oxide (NO) Production
One of the major downstream targets of BH4 is nitric oxide (NO). BH4 serves as an essential cofactor for both constitutive and inducible nitric oxide synthase (iNOS) enzymes. Moreover, along with BH4, elevated BH2 levels in ME + OI plasma samples suggest that ME + OI pathogenesis may be associated with the dysregulation of NO synthesis. Since the activation of iNOS and subsequent production of NO plays a critical role in inflammation, next we wanted to study the functional role of plasma-derived BH4 in the induction of inflammatory response in human microglial cells. To explore that possibility, we performed a plasma supplementation study followed by the analysis of NO synthesis by immunofluorescence study of iNOS and fluorimetric nitrite production assay in our cell culture model. Myeloid progenitor cell-derived cells such as microglial cells and macrophages are major sources of the iNOS enzyme. Therefore, C20 human microglial cells were included in the induction of the iNOS study. Our fluorimetric NO measurement assay demonstrated that 10 µM R5P strongly induced nitrite production in C20 microglia under hypoxic conditions (Figure 7(A)). To study the exclusive role of plasma-derived BH4, at first, the endogenous production of BH4 needs to be nullified. To attenuate PPP-induced expression of endogenous BH4, we treated microglial cells with taldo1 siRNA followed by the treatment with 25 µM R5P under an induced-hypoxia condition. Interestingly, a dual immunostaining analysis of TALDO1 and iNOS (Figure 7(B)) revealed that R5P-stimulated iNOS expression was significantly attenuated by the taldo1-siRNA treatment. Next, taldo1 siRNA-transfected and R5P-treated microglial cells were incubated with 50 µL plasma samples of ME + OI subjects and healthy controls (HC) (n = 5) under an induced hypoxia condition. ME + OI Plasma samples with high BH4 (>15 pmol/mg) and HC samples with low BH4 (<5 pmol/mg) were selected for the study. Accordingly, dual immunofluorescence analysis of TALDO1 and iNOS demonstrated that supplementation of 5% plasma samples of ME + OI, but not HC subjects, restored the expression of iNOS in R5P-treated and taldo1 siRNA-transfected C20 microglial cells under hypoxic condition. The result was further corroborated by a fluorimetric nitric oxide production assay (Figure 7(C)), in which the taldo1 siRNA was found to strongly ameliorate the production of NO in R5P-treated microglial cells. Nevertheless, the supplementation of ME + OI plasma restored the NO production in taldo1-silenced microglial cells. Functional role of plasma-derived BH4 in inducing iNOS in C20 microglial cells under hypoxic conditions. C20 microglial cells were transfected with taldo1 siRNA followed by the treatment with 10 μM R5P in a serum-free DMEM media along with HC (n = 5) and ME plasma (n = 5) at 5% (v/v) under hypoxic condition for 24 hrs. (A) after 24 hrs, fluorimetric assessment of total NO (exogenous and endogenous nitrite) was performed in C20 microglial cells treated with increasing doses of R5P under regular (green bars) and hypoxic (red bars) conditions. The significance of the mean between groups was assessed by two-way ANOVA resulting ***P < 0.001 between hypoxia control and hypoxia+10 μM R5P-treated group. (B) Immunofluorescence analysis of TALDO1 (green) and iNOS (red) was performed in taldo1 siRNA-transfected, R5P-treated, and ME + OI or HC plasma-stimulated C20 microglial cells and imaged in a fluorescence microscope. DAPI staining was adopted to stain nuclei. HCPL = HC plasma and MEPL = ME Plasma. (C) Total NO was quantified in taldo1 siRNA-transfected and 10 μM R5P-treated C20 microglial cells after 24 hrs of incubation under hypoxia conditions. A one-way ANOVA (independent variable: R5P treatment) was performed to test the significance of the mean between groups at **P < 0.01 and ****P < .0001. Ns = no significance. (D) A parametric Pearson correlation study between BH4 levels and NO-inducing abilities was performed using GraphPad Prism software with n = 10 ME + OI plasma and n = 10 HC plasma samples. Correlation was analyzed at ***P < .001 and r2 = 0.81. Results were confirmed after 3 different experiments.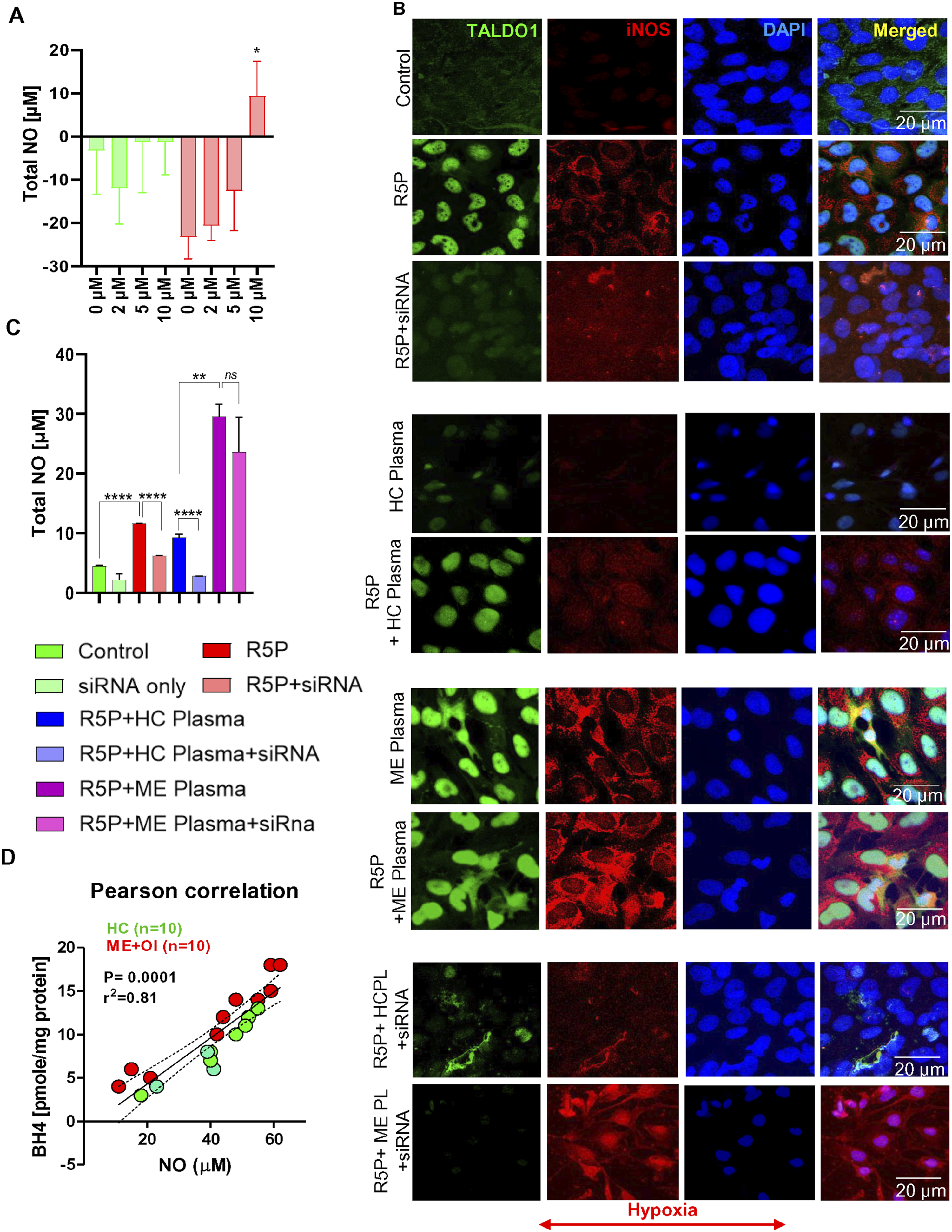
Next, we correlated the plasma levels of BH4 with the NO production ability of ME + OI (n = 10) and HC plasma (n = 10) by a parametric Pearson correlation study (Figure 7(D)). Interestingly, we observed a strong positive correlation (r2 = 0.81 and ****P < .0001) between NO and BH4 suggesting that elevated BH4 in ME + OI plasma may directly contribute to the induction of iNOS and the production of nitric oxide.
Taken together, our results suggest that plasma-derived BH4 is the outcome of induced non-oxidative PPP in ME + OI subjects and that elevated biopterins contribute directly to the induction of inflammatory response in microglial cells via upregulation of iNOS.
Materials and Methods
Reagents, Cell Lines, Antibodies, and Kits
The C20 human microglial cell line was received from Prof. L.A.A. (UWM). Primers and antibodies were purchased from Thermofisher. Antibodies such as rabbit anti-GCH1 primary (Cat#28501-1-AP), mouse anti-TALDO1(Cat#MA5-27220), rabbit anti-IMPDH2(Cat # PA527519), rabbit anti GMPS (Cat# PA565452)rabbit anti-β actin (Cat#MA5-32479), and rabbit anti-iNOS (Cat#PA1-036) antibodies were used for immunofluorescence (1:200-1:400) and immunoblot studies (1:250-1:500). Taldo1 siRNA (Cat#AM16708) was validated and commercially sold by Thermofisher. ELISA kits for quantitative estimations of BH4 (Cat# MBS733839), BH2 (Cat # MBS2568027), TALDO1 (Cat# MBS2022672), TK (Cat# MBS2024429), GTPCH1 (Cat # MBS2890723), DHPR (Cat# MBS765719), and PRPP (Cat# MBS749817) were purchased from My BioSource, INC (San Diego, CA). IRDye800-conjugated anti-rabbit and anti-mouse antibodies were obtained from LI-COR Biosciences (Lincoln, NE).
Acquisition of Human Samples
Sample cohort used in this study.
Cell Culture Under Hypoxic Conditions
The hypoxia incubator chamber (Cat# 27310) was purchased from STEMCELL Technologies. The hypoxia chamber is connected to a gas tank filled with a mixture of 85%N2 +10% CO2+ 5%O2 via silicone tubing. After treatment, the culture plate was placed on the tray inside the hypoxia chamber, sealed, turned on the gas at a flow rate of 20 L/min for 4 minutes, and then tubes were locked to stop the diffusion of gas. After 24 hrs plate was taken out and cells were assayed for different metabolic studies.
mRNA Array and Primers
List of genes, primers, and product length.
siRNA Transfection Study
C20 microglial cells were cultured in a 6-well plate. Once reached 75% confluency, cells were transfected with 0.25 µg of taldo1 siRNA (Thermofisher) via Lipofectamine™ 3000 transfection reagent as per the manufacturer’s protocol. After 24 hrs, cells were starved with serum-free media, treated with 10 μM R5P, and placed in a hypoxia chamber supplied with 85%N2: 10% CO2:5% O2 gas blend for additional 24 hrs to induce non-oxidative PPP, and then processed for subsequent enzymic and immunoassays.
Immunoblot Assay
Immunoblot assay was performed as described before. 33 Briefly, cells and tissue lysates were mixed with 5X Lamelli buffer, electrophoresed in 4%-12% Tris-Glycine gel in tris-glycine SDS running buffer, transferred in a nitrocellulose membrane (P/N 926-31090; Li-Cor Biosciences, Lincoln, NE) in tris-glycine transfer buffer, incubated with appropriate primary antibodies overnight under shaking condition, labeled with IRDye700 and 800-tagged secondary antibodies for 2 hrs at r.t in an orbital shaker, and then imaged in Odyssey Sa imager at 200 µm resolution and focus offset 3.
Immunofluorescence Study
Immunofluorescence Study was performed as described elsewhere. 33 Briefly, cells were treated with different doses of R5P under regular and hypoxic conditions with or without taldo1 siRNA based on experimental requirements. After that,
ELISA
BH4 competitive ELISA (MyBioSource; Cat# MBS733839) was performed as described previously. BH2 (MyBioSource; Cat# MBS2568027), TALDO1 (MyBioSource; Cat# MBS2022672), TK (MyBioSource; Cat# MBS2024429), GTPCH1 (MyBioSource; Cat# MBS2890723), DHPR (MyBioSource; Cat# MBS765719) and G6PDH (Abcam; Cat# ab107923) ELISA were performed as per manufacturer’s instruction. Briefly, a commercially purchased 96-well strip plate was pre-coated with primary antibody, followed by incubation with cell lysate and plasma mixed with biotinylated detection antibodies for 24 hrs at 4°C, washed with 1X TBS (or 1 X Wash buffer) for 3 times, incubated with HRP-conjugated antibody, and finally developed with TMB substrate. The reaction was stopped with an acidic stop solution and read at 450 nm wavelength in an Enspire (PerkinElmer) universal plate reader.
Enzyme Activity Assay
G6PDH Assay
Glucose 6 phosphate dehydrogenase (G6PDH) assay – Blood samples were collected and layered on LymphoprepTM (Stem Cell Technologies) solution at Sepmate-15 tubes as recommended by the manufacturer. Subsequent centrifugation at 1000xG for 10 minutes was applied to isolate plasma/serum (top layer) and PBMCs (interface). PBMC was collected, lysed with cell lysis buffer (supplied with the kit), and immediately stored at −80°C. 50 µL of cell lysate was assayed for the Glucose 6 phosphate dehydrogenase activity kit (Fluorometric) (Abcam; ab#176722) following the manufacturer’s instructions. Briefly, the reaction was set by mixing the sample and the reaction mixture (assay buffer + NADP stock solution + enzyme probe) in a 96-well plate. The absorbance of each sample was monitored by fluorescence increase at Ex/Em = 540/590 nm on an Enspire (PerkinElmer)microplate reader in kinetic mode, every 5-10 minutes, for 1 hr at room temperature, protected from light.
TK Assay
TK assay was performed as per the manufacturer’s protocol (Cat# ab273310). Fifty microliters of cell lysate and plasma (<0.6 μg protein) were loaded in a 96-well plate and then conjugated with the 50 μL of the reaction mixture (Assay buffer + substrate + Developer + enzyme + picoprobe). After that, the reaction progression was started recording kinetically with Ex/Em = 535/587 nm at 30-second intervals for 45 minutes at 37°C.
NADP/NADPH assay
Fifty microliters of PBMC lysate was assessed for NADP/NADPH assay as per the manufacturer’s instruction (ab176724).
Statistical Analyses
For a 99% confidence interval and 0.05 significance, our sample size calculation is n = z2×p (1− p)/ε 2 = 1.282 × 0.99 (1− 0.99)/0.052 = 7. Z is the z score, which is 1.28 for power 0.8; p is the population proportion. For a 99% confidence interval, p will be 0.99; ε is the margin of error = 0.05. Therefore, throughout the study, we selected at least n = 7 per group when comparing results between HC and ME groups.
Discussion
The role of BH4 in the pathogenesis of ME + OI has recently been emphasized since we reported that the plasma samples of ME + OI subjects had elevated BH4. The result is unexpected as the deficiency, but not the sufficiency, of BH4 is plausible and frequently reported in various metabolic disorders. BH4 sufficiency was never reported in any metabolic disorder before. The synthesis and metabolism of BH4 are tightly regulated and conserved in a healthy individual. BH4 deficiency happens primarily due to rare genetic mutations or partly due to metabolic impairment. Its deficiency is manifested by several metabolic alterations such as phenylketonuria, loss of dopamine synthesis, and hypocitrulinemia. All these abnormalities can be efficiently detected by a comprehensive analysis of the blood amino acids panel. Interestingly, these tests never provided any conclusive evidence of BH4 deficiency in ME/CFS subjects. In contrast, while measuring BH4 in ME/CFS subjects, our previous study identified that plasma samples of ME/CFS subjects had elevated BH4. In our current study, we explored the molecular mechanism of BH4 upregulation in ME + OI subjects. Several lines of our manuscript demonstrated that the augmentation of the non-oxidative metabolism of glucose via PPP is critical in the pathogenesis of ME + OI.
Augmentation of Non-Oxidative PPP is Pivotal in the Upregulation of BH4
First, our microarray followed by a real-time mRNA validation study revealed that expressions of genes encoding key enzymes of PPP such as glucose-6-phosphate dehydrogenase (6gpdh), 6-phosphogluconolatonase (6pgl), and transaldolase (taldo1) were strongly upregulated in ME + OI subjects. Upregulations of these genes were coupled with the reduction of oxidative glycolytic genes such as hexokinase (hk) and glyceraldehyde-3-phosphate dehydrogenase (gapdh). Along with these downregulated genes, we also observed the upregulation of the non-oxidative glycolytic gene lactate dehydrogenase (ldh) suggesting that ME + OI pathogenesis might be associated with the augmentation of a non-oxidative mode of glucose metabolism. Second, our ELISA analyses further confirmed that glucose-6-phosphate dehydrogenase (g6pdh), a rate-limiting enzyme of PPP, was strongly upregulated in ME + OI subjects compared to age-/gender-matched healthy controls indicating that the augmentation of PPP may be critical in the pathogenesis of OI in ME/CFS. Third, the protein expression of TALDO-1, the critical enzyme of non-oxidative PPP, had also been found to be strongly upregulated in the PBMC lysates of ME + OI subjects compared to the control. Finally, the enzymic activity of Transketolase (TK) was also found to be strongly elevated in the PBMCs of ME + OI patients.
A Novel Cell Culture Model to Study BH4 Metabolism in ME + OI Patients
Suppression of aerobic glucose metabolism,
34
reduced mitochondrial oxygen consumption,
35
and diminished mitochondrial energy production
36
in ME/CFS patients indicate that the anaerobic glucose metabolism could play a critical role in the pathogenesis of ME/CFS patients. Moreover, our results also suggested that the expressions of several genes such as lactate dehydrogenase (ldh), transaldolase1 (taldo1), transketolase (tk) were strongly upregulated in ME + OI subjects. Induction of ldh gene indicates the augmentation of anaerobic glycolysis, whereas upregulations of taldo and tk genes suggest the induction of non-oxidative PPP. Collectively, these results suggest that the implementation of a reduced oxygenated environment in the cell could stimulate a reductive metabolic condition, which upregulated expressions of ldh, taldo, and tk in microglial cells similar to ME + OI subjects. To achieve that metabolic condition, we adopted the following strategies. First, we implemented a hypoxic environment that augments a reductive potential in the microglia causing reversible enzymes TALDO and TK to increase the endogenous flux of R5P. Second, the application of exogenous R5P in cells conditioned with reduced oxygen supply was adopted. R5P is the precursor molecule of anaerobic PPP, purine nucleotide GTP, and BH4. Accordingly, we designed a cell culture strategy in which C20 human microglial cells were first treated with 10 μM R5P and then immediately sealed in a hypoxia chamber supplied with a gas mixture of 85% N2, 10% CO2, and 5% O2. Our subsequent analyses indicated that the R5P treatment in hypoxic conditions upregulated the expression of TALDO1 and induced the enzymic activity of TK. Interestingly, several downstream metabolic analyses indicated that the implementation of non-ox PPP triggered the expressions of purine intermediates, BH4, and other biopterin metabolites in ME + OI subjects. First, the level of PRPP was found to be elevated in microglial cells when treated with R5P under hypoxic conditions. Second, Expressions of key enzymes of purine biosynthesis such as IMPDH2 and GMPS were also upregulated under similar conditions. Third, an ELISA analysis of GCH1, a rate-limiting enzyme of BH4 biosynthesis, revealed that the implementation of hypoxia condition in 10 μM R5P-treated microglia strongly induced the expression of GCH1. Fourth, the dual immunostaining analyses confirmed that the activation of anaerobic PPP, as indicated by TALDO1 upregulation, indeed upregulated GCH1 expression in C20 microglia. Fifth, the Quantification of BH4 in the cell lysate of C20 microglial cells by a competitive ELISA assay directly confirmed that the induction of anaerobic PPP truly upregulated BH4 level. Sixth, silencing the endogenous expression of TALDO1 by siRNA strongly downregulated the expression of GCH1, and finally
BH4 Upregulation: Is it a Cause or Result of Metabolic Impairment in ME + OI Subjects?
BH4 is an important cofactor in many essential metabolic pathways. Upregulation of BH4 is beneficial in reducing inflammation and stress in metabolic diseases ranging from diabetes to cancer, neuroinflammatory diseases to rare genetic disorders. Therefore, it is puzzling to decode the role of upregulated BH4 in the pathogenesis of ME + OI. At first, we asked if the upregulation of BH4 was the central and independent mechanism that contributed to the pathogenesis of ME + OI or if it was a secondary response to some other upstream metabolic impairments. Impaired glucose metabolism and the resultant deficit of mitochondrial energy are shown to be involved in ME/CFS pathogenesis. As an alternative strategy, the pentose phosphate pathway could be operative for glucose utilization. A recent study [32] showed that PPP was truly activated in ME/CFS subjects. However, our study showed that the augmented PPP did not shunt the glycolytic energy production mechanism but continued to proceed through the anaerobic stage, which might facilitate R5P to follow the purine nucleotide biosynthetic pathway for the synthesis of GTP, induced GTPCH1 enzyme activity that further caused the upregulated synthesis of BH4 (graphical summary at Figure 8). Therefore, BH4 upregulation possibly happened as a secondary event due to the augmentation of non-oxidative PPP. Graphical summary. (Red enclosure) Reported molecular mechanisms on how the impairment of glycolysis, deficiency of pyruvate transport, reduced oxygen consumption, and uncoupling of electron transport chain could contribute to the failure of energy metabolism in ME/CFS pathogenesis. (Green Enclosure) Our current research demonstrates how the alternative glucose metabolism under reduced oxygenated environment via PPP could stimulate the biopterin biosynthesis. Transaldolase (TALDO) and transketolase (TK) are 2 reversible enzymes in non-oxidative PPP maintaining the bioavailability of ribose-5-phosphate (R5P). Implementation of hypoxic conditions imposed a strong reductive potential in the cell driving the reaction equilibrium to enhance the production of R5P. The increased R5P then adopts purine biosynthetic pathway under the reduced oxygenated condition via induction of phosphoribosyl pyrophosphate (PRPP) biosynthesis leading to the formation of BH4.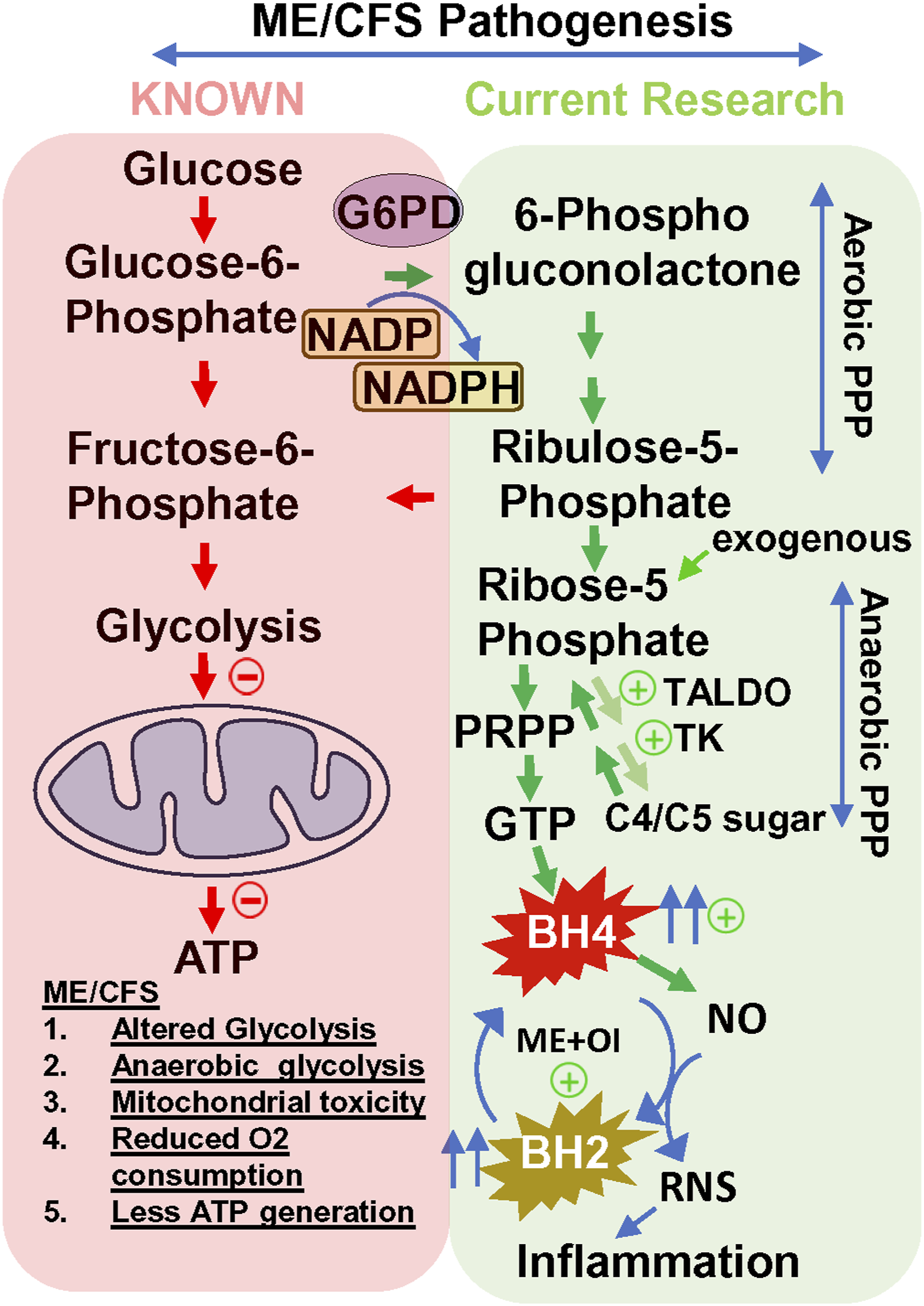
How did BH4 Contribute to the Pathogenesis of ME + OI?
Our study showed that the augmentation of anaerobic PPP and the resultant upregulation of BH4 could stimulate iNOS in microglial cells. First, a fluorimetric NO measurement assay revealed that the activation of anaerobic PPP by treating microglial cells with 10 μM R5P under hypoxic conditions stimulated NO. Interestingly, under similar conditions, we observed a strong upregulation of BH4. Second, A dual immunofluorescence study also revealed that the induction of anaerobic PPP as indicated by enhanced TALDO1 expression also upregulated iNOS expression, and the knocking down of TALDO1 expression by siRNA also ameliorated iNOS expression. Interestingly, under similar conditions, we observed that taldo1 siRNA also inhibited BH4 expression in C20 microglial cells. Third, the exogenous addition of ME + OI serum samples with high BH4 (>15 pmol/mg) induced iNOS expression and NO production, whereas administration of HC serum samples with low BH4 (<5 pmol/mg protein) was unable to induce the same. Finally, A parametric correlation statistical analysis between the levels of BH4 and NO production capacity in a total of 20 serum samples (n = 10 ME + OI and n = 10 HC) demonstrated that the elevated BH4 could be directly linked to the induced NO production under hypoxic conditions. Is that NO pathologically relevant? Interestingly, we also observed a strong upregulation of BH2 in ME + OI serum samples. Implementing non-oxidative PPP also upregulated BH2 in C20 microglia. Studies suggest 37 that the accelerated conversion of BH4 to BH2 could uncouple physiological NO production and induce cellular stress. Therefore, based on our analyses, a significant proportion of the elevated BH4 may be converted to BH2 in ME + OI subjects and directly correlated with the increased iNOS activation and NO production in microglial cells.
Future Direction
The metabolic role of anaerobic PPP is a complex mechanism, which is regulated by other pathways of glucose metabolism. Therefore, a high-resolution metabolomics study should be pursued in a large sample cohort followed by machine learning-based network analyses of all metabolomes will confirm the role of PPP-induced BH4 in ME + OI pathogenesis. In our future goal, we will use our biobank of ME + OI samples to perform the metabolomic study in-house (UWM) State-of-the-art Analytical Instrumentation Laboratory and Research Core (SAILRAC) facility (https://sites.uwm.edu/sailarc/).
Supplemental Material
Supplemental Material - Dysregulation of tetrahydrobiopterin metabolism in ME/CFS by pentose phosphate pathway
Supplemental Material for Dysregulation of tetrahydrobiopterin metabolism in ME/CFS by pentose phosphate pathway by Sarojini Bulbule, Carl Gunnar Gottschalk, Molly E. Drosen, Daniel Peterson, Leggy A. Arnold and Avik Roy in Journal of Central Nervous System Disease
Footnotes
Acknowledgements
We are grateful to Dr Maureen Hanson of Cornell University for her valuable support in the acquisition of PAXgene RNA samples of ME + OI subjects.
Author Contributions
A.R. conceived the idea. S.B., C.G.G., M.E.D., and A.R. designed, performed research, and wrote the manuscript. C.G.G. was in charge of gathering human samples and keeping subjects’ records. D.P. is a clinical PI, who was in charge of collecting biospecimen, questionnaires, and IRB approval. L.A. provided resources and facilitated the collaborative research between UWM and Simmaron. All authors have read and agreed to the published version of the manuscript.
Ethical Statement
Declaration of Conflicting Interest
The author(s) declared the following potential conflicts of interest with respect to the research, authorship, and/or publication of this article: A.R., C.G.G., S.B., M.E.D., and D.P. are employees of Simmaron Research INC, a 501C non-profit research organization. The authors declare no conflict of interest.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by Simmaron Research Inc., a non-profit 501C research organization, Incline Village, NV 89451, as a result of funds from NIH R21 grant (R21NS129021-01A1) to AR, Ramsay Award 2023 to AR by Solve ME initiative and donation from Foundation Hesse Sibylla, Quebec, QC, Canada.
Research Data Declaration
Data and biospecimens used in the current project will be shared with researchers around the world upon request.
Data Availability Statement
There is no electronic datasheet associated with this paper. No data in an electronic repository.
Supplemental Material
Supplemental material for this article is available online.
Abbreviations
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.