
Abstract
MNGIE (Mitochondrial Neurogastrointestinal Encephalomyopathy) is an ultra-rare autosomal recessive disorder that leads to mutations in the nuclear genes encoding thymidine phosphorylase. Symptoms include gastrointestinal dysmotility, cachexia, ptosis, external ophthalmoplegia, sensorimotor neuropathy and asymptomatic leukoencephalopathy. We describe the first case of MNGIE with meningoencephalitis that ultimately led to a familial diagnosis ending a diagnostic odyssey. We retrospectively reviewed the electronic medical records and sent whole exome sequencing for the index case and his family members. We report the variant c.877T>C p.(Cys293Arg) found in TYMP gene in all affected siblings showed typical clinical manifestations related to MNGIE. To the best of our knowledge, this is not described in the literature nor in the population databases dbSNP (Single Nucleotide Polymorphism Database) and gnomAD (Genome Aggregation Database). Additionally, it is located in a highly conserved residue and the bioinformatic analysis suggests it is most probably deleterious. Moreover, we estimated 550 number of cases of MNGIE (including 5 cases in this study) after performing an extensive search in the literature across 3 databases from 1983-2023. In addition, we identified 44 patients with MNGIE-like phenotype in genes other than TYMP. MNGIE-like phenotype affects POLG1, RRM2B, LIG3, RRM1, MTTV1, and MT-RNR1 genes.
Plain language summary
A rare neurological presentation unravels a family’s medical mystery after years of no diagnosis: MNGIE is a rare disease caused by changes in a gene that cause deficiency in an enzyme called thymidine phosphorylase. Patients complain of significant weight loss, tingling and numbness in their extremities, muscle weakness, digestive issues and drooping eyelids. We encountered a patient with symptoms and signs of inflammation of the brain and it's protective lining. However, laboratory tests were inconclusive whilst his condition kept deteriorating. A genetic analysis revealed a new mutation not described in the literature before. This has also helped to diagnose the entire family after years of not receiving an answer regarding their symptoms. We also found 550 cases of MNGIE published in the scientific literature from 1983 to 2023. This case highlights the importance of taking a family’s entire family history and genetic testing to solve complex medical cases.
Keywords
Introduction
MNGIE (Mitochondrial Neurogastrointestinal Encephalomyopathy) is a rare multi-system disorder with a prevalence of 1-9 per 1 million.
1
It is caused by a mutation in the TYMP gene and is inherited in an autosomal recessive manner. The identified gene locus is on 22q13.33 and the deficient enzyme is Thymidine Phosphorylase (TP). This affects de novo pyrimidine dNTP (deoxynucleotide triphosphate) metabolism leading to cytotoxic accumulation of thymidine and deoxyuridine in organs throughout the body. It presents within the second to 3rd decade of life.
2
The classic clinical manifestations are summarized in Figure 1. Due to the clinical heterogeneity and rarity of the disorder, patients are misdiagnosed which leads to a delay in diagnosis.
3
This prevents timely administration of therapies that could possibly improve the overall clinical outcome. Classical clinical features in MNGIE (mitochondrial neuro-gastrointestinal encephalomyopathy).
We present an unusual case of meningoencephalitis in a previously undiagnosed patient that led to a familial diagnosis of MNGIE. To date, there has been no report of meningoencephalitis as an atypical manifestation in MNGIE. Atypical radiological findings are described as well. In addition, 114 mutations have been described in the literature. 4 This case has a novel mutation, which might explain the unusual clinical manifestations. We also performed a literature search to identify number of reported cases of MNGIE till December 2023.
Case description
Case 1 (proband)
A 35-year-old cachectic male, was accompanied to the ER (emergency room) with a meningoencephalitis-like picture with headache, nausea and vomiting for 4 days. In addition, he had several spikes of fever, confusion and personality changes. Initial CT (computed tomography) scan of the brain was normal. He was started on empirical ceftriaxone, acyclovir and vancomycin.
A CSF (cerebrospinal fluid) lumbar puncture was ordered which revealed clear CSF, with pleocytosis of 50 cells/μl (lymphocytes 38%, monocytes 32%, neutrophils 30%), RBC (red blood cell) count of 250 cells/μl, glucose 3.2 mmol/L and protein .80 g/dl. No organisms were isolated by microbiological culture or viral PCR (polymerase chain reaction) multiplex. The patient became unresponsive 2 days later and was found to have non-convulsive status epilepticus along with a hypodensity and edema in the left temporal lobe on repeated CT scan of the brain. He was treated accordingly and despite the resolution of the electrographic convulsions, the patient did not show any noticeable improvement in his level of consciousness.
Past medical history revealed on and off diarrhea and inability to tolerate fasting in the month of Ramadan along with weight loss. Given the long-standing history of anorexia and weight loss, he was started tentatively on intravenous immunoglobulin (IVIg) to cover for autoimmune/paraneoplastic limbic encephalitis. Following that, he showed improvement in sensorium and was extubated. Autoimmune and paraneoplastic encephalitis panel were sent for Anti-Hu, Anti-CV2, Anti-Amphiphysin, Anti-Yo, Anti-Tr, Anti-ribosomal p, Anti-voltage gated calcium channel antibodies, Anti-NMDAR (N-methyl D-aspartate receptor), anti-AMPAR (α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptor), anti-DPPX (dipeptidyl-peptidase–like protein 6), anti-LGI1 (leucine-rich-glioma-inactivated 1), anti-GAD65 (65 kDa isoform of glutamic acid decarboxylase), anti-Ma, anti-Ta, anti-Sox1 (SRY-related HMG-box) which returned negative. Infectious panel PCR (polymerase chain reaction) multiplex for bacterial (E.Coli K1, Haemophilus Influenzae, Listeria monocytogenes, Neisseria meningitidis, Streptococcus agalactiae, Streptococcus pneumoniae), viral (cytomegalovirus, enterovirus, herpes simple virus 1 and 2, human herpesvirus 6, human parechovirus, varicella zoster virus), fungal (Cryptococcus neoformans), CSF Mycobacterium tuberculosis MTB PCR, CSF VDRL (venereal disease research laboratory) were negative as well.
MRI (magnetic resonance imaging) brain (Figure 2(a)-(g)) shows diffuse relatively symmetrical altered signal intensity involving bilateral frontal and temporal lobes seen as T2 (2(a)-(b)) and FLAIR (fluid attenuated inversion recovery) hyperintensity (2(c)-(d)) showing gyral thickening/edema and corresponding hypo intensity on T1 weighted (2(d)) images with poor delineation of gray-white matter differentiation. Changes are more extensive involving left temporal lobe and bilateral hippocampal lobes. There was also diffuse leptomeningeal enhancement in the post contrast images (2e). No diffusion restriction was noted (2(g)-(h)). (a-h) demonstrates diffuse relatively symmetrical altered signal intensity involving bilateral frontal and temporal lobes - seen as T2 (a-b) and FLAIR (c-d) hyperintensity showing gyral thickening/edema and corresponding hypo intensity on T1 (e) weighted images with poor delineation of gray-white matter differentiation. Changes are more extensive involving left temporal lobe and bilateral hippocampal lobes. No true restricted diffusion (g-h). Leptomeningeal enhancement (f)
There is history of consanguinity amongst his parents with 13 siblings. No symptoms were reported by both parents. However, family history revealed a history of abdominal pain in 4 of his siblings along with diplopia and ophthalmoplegia. One sibling was given the provisional diagnosis of MELAS (mitochondrial encephalomyopathy, lactic acidosis and stroke-like episodes) on clinical grounds; no genetic testing was ordered. Another sibling was given a diagnosis of polyneuropathy with clinical suspicion for Charcot-Marie-Tooth disease pending the genetic testing results.
Routine laboratory testing was unremarkable apart from thrombocytopenia with platelet count 137,000 cells/mm3 (150,000-450,000 cells/mm3) and consistently elevated lactic acid levels – 4.5 mmol/L (reference range .5-2.2). Considering logistical difficulties in sending samples abroad within 24 hours of collection and analyzing samples on ice we could not perform TP enzyme activity or nucleoside level analysis. 5
A serendipitous review of his past family history through electronic medical records revealed a loss to follow-up appointment for one of his siblings. He presented with gastrointestinal manifestations with genetic testing revealing homozygous TYMP gene mutation. Proband was subsequently tested using whole exome sequencing revealing the variant c.877 T>C p.(Cys293Arg) causing an amino acid change from Cys to Arg at position 293 and it was detected in apparent homozygosity in the TYMP gene at chromosome 22. A diagnosis of MNGIE was subsequently made. His siblings were later genetically tested revealing a positive mutation in three of his siblings. Intra-familial phenotypic similarities were observed.
The patient was offered the experimental therapy of platelet transfusion that led to transient improvement in level of consciousness. 6 However, unfortunately he couldn’t be weaned off the ventilator due to respiratory distress and developed multiple hospital acquired infections which led to his demise.
Sibling 1
A 52-year-old woman presented with anorexia, abdominal distension, post-prandial abdominal pain, intermittent sporadic diarrhea, and fatigue since the age of 36 years. She was always underweight and currently has a BMI (body mass index) of 12.6 (weight 29.8 kilograms). Additional symptoms include chronic fatigue, proximal upper and lower limb weakness. This manifested with difficulty in climbing stairs and combing her hair. She had no bulbar symptoms or sensory abnormalities. At 38 years of age, she developed diplopia and progressive ptosis. An impression of progressive external ophthalmoplegia was made. On examination, proximal muscle weakness grade 4+ medical research council (MRC scale) with diminished reflexes 1+. Nerve conduction study (NCS) showed sensory motor neuropathy. Repetitive nerve stimulation showed no decrement and her Tensilon test was negative. Her vasculitic screen and anti-acetylcholine receptor antibodies were negative as well.
The afore-mentioned symptoms, in addition, to syncopal attacks led to a neurology consultation, where magnetic resonance imaging revealed incidental leukoencephalopathy. She was prescribed a short course of corticosteroids with minimal clinical benefit. A muscle biopsy was obtained and was suggestive of mitochondrial disease, however, no genetic studies were done at that time. A preliminary impression of suspected MELAS was made. She was placed on a mitochondrial cocktail of biotin, co-enzyme Q10 and carnitine and reported a salutary benefit of increased energy and increase in motor strength.
At 45 years of age, she reports that her speech started lacking cadence and she had trouble in articulation for which she undergoes speech therapy at the present. Moreover, she has symptoms of dysphagia and sialorrhea. She mentions symptoms of hearing loss necessitating audiological evaluation. She is currently on dietitian recommended protein supplements.
Genetic testing after proband’s positive result confirmed a diagnosis of MNGIE. MRI imaging (Figure 3(a)-(c)) revealed diffuse periventricular and subcortical white matter hyperintensity with extension towards the external capsule and posterior fossa white matter. She was offered an option of liver transplantation and peritoneal dialysis as treatment options, however she tentatively declined. (a-c) demonstrates bilateral diffuse periventricular and subcortical white matter T2 and FLAIR hyperintensities with extension toward the external capsules and posterior fossa white matter and faint bright signal intensity within the dorsal thalami bilaterally.
Sibling 2
This 42-year-old male developed ptosis and fatigue at the age of 31 along with anorexia, abdominal distension and postprandial abdominal pain. He currently weighs 48 kilograms and has a height of 180 centimeters (BMI – 14.81). He reports developing peripheral neuropathy in both his lower extremities with symptoms of numbness, cold extremities without color changes and a neuropathic itch. He mentions developing gait imbalance one year ago along with erectile dysfunction (ED). All investigations for ED were normal and he is on Tadalafil.
MRI study obtained revealed ill-defined bilateral symmetrical and confluent hyperintensities involving supratentorial periventricular and subcortical white matter in fronto-parietal lobes as shown in Figure 4(a) and (b). (a and b) - demonstrates bilateral symmetrical T2 and FLAIR hyperintensities involving mainly supratentorial periventricular and subcortical white matter in fronto-parietal lobes.
He received a positive diagnosis upon genetic testing of the family following the proband’s diagnosis. He was placed on the mitochondrial cocktail. He reports no improvement in symptoms and feels that his symptoms are becoming progressively worse especially neuropathy in the left lower limb. He developed new onset palpitations pending a cardiology follow-up appointment. He was offered treatment option of liver transplant and peritoneal dialysis as well but has tentatively declined.
Sibling 3
A 34-year-old male with a BMI of 14.1 (weight – 41 kilograms, height 170 centimeters) reports developing progressive weight loss in addition to abdominal pain and vomiting at the age of 28. Other GI (gastrointestinal) manifestations include alternating diarrhea with constipation, bloating, painful defecation and gastroesophageal reflux were also reported.
He developed symptoms of peripheral neuropathy at the age of 30. Symptoms included neuropathic pain in all extremities, numbness, and cold extremities. He also reports additional nocturnal cramps. He reports no ptosis or opthalmoparesis
He received a positive diagnosis following the genetic testing of the family. He reports improvement in GI symptoms following mitochondrial cocktail. He reports being able to fast in Ramadan now and improved energy and performance at work.
MRI of the brain (Figure 5(a)-(d)) revealed ill-defined bilateral symmetrical and confluent T2 and FLAIR (Fluid Attenuated Inversion Recovery) hyperintensities involving supra and infratentorial white matter as well as patchy involvement of the basal ganglia, thalami, external capsule, dorsal pons. Treatment options were discussed including peritoneal dialysis and liver transplantation. He agreed for the option of transplantation, awaiting a possible donor. a-f) - illustrates ill-defined bilateral symmetrical and confluent T2 (5a) and FLAIR (5d) hyperintensities involving periventricular and subcortical white matter in fronto-parietal lobes and patchy similar hyperintensities in bilateral cerebellar white matter (5c).
Sibling 4
A 33-year-old male with a BMI of 10.98 (weight – 36.4 kilograms, height 182 centimeters) developed a history of chronic diarrhea starting 4 years ago. It was watery in consistency and he would have 5 episodes per day. Other GI manifestations include abdominal cramps and nausea. He had no fever or bloody stools with the episodes. He also had a history of dysphagia to both solids and liquids causing severe decrease in weight. On examination, he had stable vital signs, no pallor or icterus, wet mucous membranes, no organomegaly and no tenderness on abdominal examination. The remainder of the systemic examination was normal. CT scan of the abdomen and thorax was done in 2022 to rule out malignancy. It revealed significant circumferential wall thickening and enhancement involving the entire bowel loops predominantly the small bowel loops with associated ascites. An impression of active inflammatory bowel disease mainly Crohn’s disease with less likelihood of an infectious etiology. No masses or collections were noted. Scattered areas of ground glass haziness and centrilobular nodules mainly in the right lower lobe suggestive of an infectious process. He underwent colonoscopy evaluation for the chronic diarrhea and few diverticuli were observed. Upper gastrointestinal esophageal gastroduodenoscopy done revealed mild esophagitis, normal gastric study, small ulcer in the first part of the duodenum. A biopsy was obtained, duodenitis was observed in the second portion of the duodenum as well.
He developed symptoms of peripheral neuropathy as well, proximal lower limb muscle weakness causing motor weakness (difficulty in climbing stairs and driving vehicles), coldness in peripheral extremities and numbness. Concurrently, he had hypokalemia which was believed to be the cause of symptoms and he was repleted. He could fast during Ramadan with no issues and had no ptosis or ophthalmoplegia/ophthalmoparesis. He reported asthenia and fatigue. MRI brain taken in 2022 (Figure 6(A)-(F)) revealed ill-defined bilateral symmetrical and confluent T2 and FLAIR hyperintensities involving periventricular and subcortical white matter in fronto-parietal lobes and patchy similar hyperintensities in bilateral cerebellar white matter. Mild volume loss changes were noted as well. Features were consistent with leukoencephalopathy related to metabolic disorder. Nerve conduction study (NCS) showed a pattern of mixed axonal and demyelinating neuropathy. A genetic panel for Charcot-Marie Tooth disease was sent which was negative. (a-f) - Ill defined bilateral symmetrical and confluent T2 and FLAIR hyperintensities involving supra and infratentorial white matter as well patchy involvement of basal ganglia, thalami, external capsule, dorsal pons. Features are consistent with leukoencephalopathy related to MNGIE.
He has a history of osteomyelitis of the foot and scalp abscess successfully treated with debridement and antibiotic coverage. He had a history of erectile dysfunction as well scheduled for penile doppler ultrasonography and hormonal profile workup.
He developed productive cough with white sputum and fever in May 2022 which progressively increased in severity. Chest examination revealed reduced air entry on the right side. An impression of right sided pneumonia was made and he was started on Amoxicillin-Clavulanate and Azithromycin empirically. He absconded from the ER and revisited after several days, with decreased oxygen saturation and loss of consciousness necessitating an ICU (intensive care unit) admission. He was intubated and was on ventilator support with norepinephrine. Chest X ray revealed worsening bilateral infiltrates and laboratory testing revealed cytopenia (leukopenia and thrombocytopenia) with metabolic acidosis. He subsequently succumbed to septic shock.
He had a genetic test done 4 years back which confirmed MNGIE diagnosis, however there was loss to follow-up. Examination of his electronic medical records led to serendipitous testing of the proband and familial diagnoses.
Pedigree of the entire family for three generations is summarized in Figure 7. Pedigree analysis showing proband and his family relations described for three generations along with pedigree key.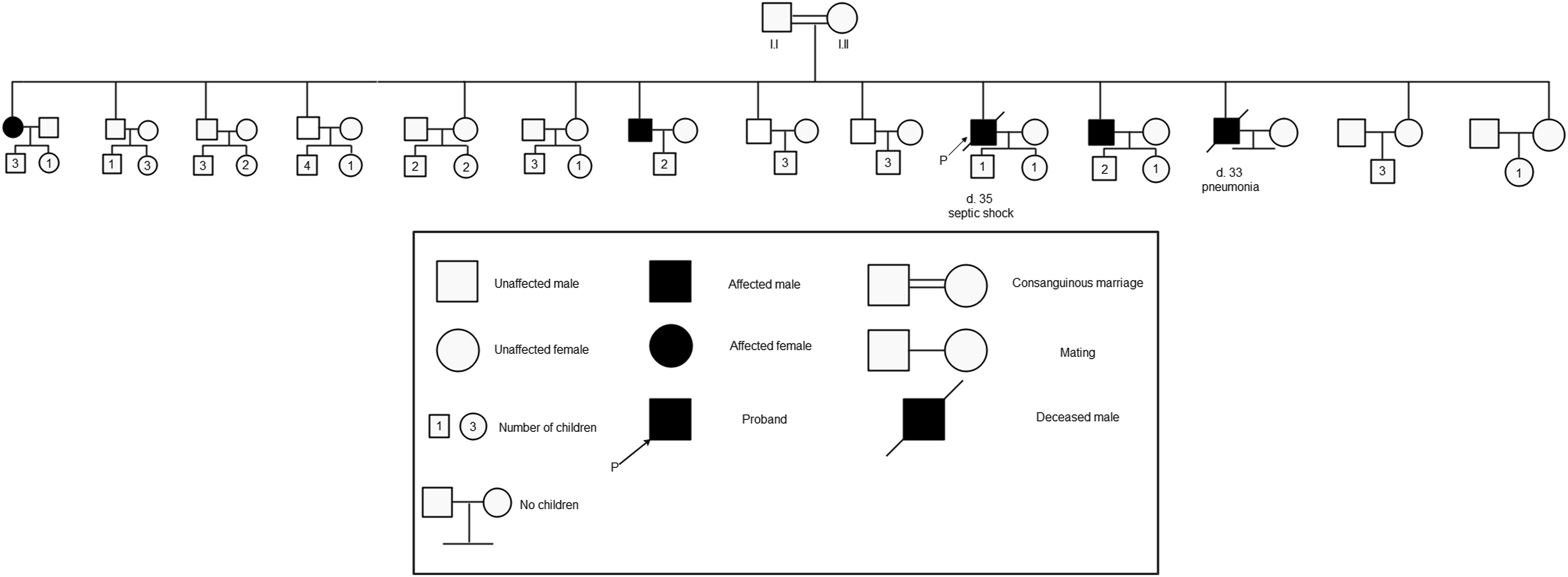
Number of cases of MNGIE
The prevalence of MNGIE is unknown. 7 In previous reports, a cohort 102 cases were identified between 1988 and 2011. 8 In ITA-MNGIE study, an Italian national and regional survey of MNGIE estimated prevalence at ∼.15 in a million. 9 Orphanet estimates the prevalence to be 1-9 per 1 million . 1 No reports were found for an estimate of MNGIE-like phenotype caused by mutations in genes other than TYMP.
The reporting of the search strategy is in accordance to PRISMA-S (Preferred Reporting Items for Systematic reviews and Meta-Analyses literature search extension) guidelines. 10 The checklist can be accessed in Supplementary file 2 (pages 31-32). We sought to identify the total number of cases of MNGIE from published papers in all languages across a 40 year period 1983-2023.
The following electronic databases were searched: MEDLINE (Ovid), Scopus (El Sevier) and Google Scholar. No databases were searched simultaneously using a single platform. Clinical trial study registries were not searched. Conference proceedings were searched and accessed using Google Scholar for unpublished abstracts. Dates and names of conference proceedings are reported in the references for MNGIE and MNGIE-like phenotype in supplementary files 1 and 2 respectively. References of earliest studies of MNGIE (1987-1994) were screened to identify additional prior studies or studies with different terminology (Polyneuropathy, Ophthalmoplegia, Leukoencephalopathy, and Intestinal Pseudo-obstruction: POLIP Syndrome; Oculogastrointestinal muscular dystrophy). For studies whose papers or abstracts could not be accessed, authors were emailed to solicit additional information. No additional information sources were used.
Full search strategy with words and Boolean terminology is summarized in page 32 of supplementary file 2. No limits or search filters were imposed during the search process. No search strategies from prior published works were adapted. A comprehensive literature search was run on 2nd August 2022 and the last date of the search rerun across all platforms was on 15th March 2023. Email alerts were also set for prospective publications until 21st December 2023. No search peer review process was utilized.
A total of 10,082 citations were retrieved from the three databases. A total of 820 duplicates were identified and removed using EndNote reference manager (version 20 by ClarivateTM) and subsequently manually. 8996 animal studies, reviews, non-clinical studies, studies reporting on the same patients, non-MNGIE search results were subsequently excluded. A total of 266 studies were perused for cases. Efforts were made not to recount cases that have been reported in multiple published papers.
In addition to the 5 cases with novel mutation described in this paper, we identified 550 cases of MNGIE across a 40 year period between 1983-2023. In addition, we identified 44 patients with MNGIE-like phenotype – 7 cases with LIG3 gene,11,12 24 cases with POLG1 gene,13-27 8 cases with RRM2B gene ,28-30 1 case with RRM1 gene, 31 1 case with MTTV gene, 32 1 case with MT-RNR1 gene, 33 heterozygous PEO1-TYMP gene mutation, 34 and 2 cases with multiple mtDNA mutations. 35
Discussion
We hereby describe a novel mutation in thymidine phosphorylase causing varying clinical manifestations, some of which have not been reported previously, in a family diagnosed with MNGIE.
The variant c.877 T>C p.(Cys293Arg) found in TYMP gene in all affected siblings showed typical clinical manifestations related to MNGIE. To the best of our knowledge, this is not described in the literature nor in the population databases dbSNP and gnomAD. Additionally, it is located in a highly conserved residue and the bioinformatic analysis suggests it is most probably deleterious. With the available information, this variant classified as a VUS (variant of uncertain significance) as per ACMG (American College of Medical Genetics and Genomics) guidelines. In the future, it would be best to have a functional study to prove its significance. As our hospital is not a research facility this cannot be done. Because of the effect of the variant on gene function and since it was found in all affected patients, we report this novel variant as disease causing.
To date, this is the first description of a case of MNGIE with radiological and laboratory diagnosis of meningoencephalitis as evidenced by the CSF results and the diffuse leptomeningeal enhancement on MRI in the index case and sibling 3. Our index case presented with a typical history of meningoencephalitis with fever, headache and personality changes which ultimately led to his diagnosis. However, a culprit pathogen was never found despite extensive testing. The patient did not show improvement after antimicrobial administration. However, there was significant improvement in sensorium after IVIG (intravenous immunoglobulin) administration. This could be attributed to the immune boost provided to fight a long standing immunodeficiency. The autoimmune/paraneoplastic panel was also negative. Sibling 3 also had a similar radiological picture but he is asymptomatic with no features of meningoencephalitis.
Notoriously, it has been known that asymptomatic leukoencephalopathy is a hallmark of MNGIE and its presence in combination with the gastrointestinal and neurological symptoms significantly narrows the diagnosis to MNGIE. 6 TP is abundantly expressed in the cells of the digestive tract and the neurons. 36 TP deficiency ultimately leads to accumulation of thymidine and deoxyuridine that leads to secondary mitochondrial DNA mutations and depletion. One may speculate that the ongoing leukoencephalopathy and dysmyelination resulting from the mutations, although reported as typically paucisymptomic, releases epitopes that trigger autoimmunity as reported in other disorders of dysmyelination such as metachromic leukodystrophy. 37
Furthermore, there is a report of a patient with MNGIE experiencing trigeminal neuralgia corresponding intrapontine trigeminal fibers lesions on imaging identical to those described in multiple sclerosis. 38 This might suggest the presence of an additional inflammatory response contributing to the pathology. Another conjecture is that the meningoencephalitis resulted from a state of immunodeficiency. 39
Some siblings had different clinical manifestations some of which were typical, whilst other had atypical ones, these differences reflect phenotypic variability between the siblings. Serum levels of dThd (deoxythymidine) and dUrd (deoxyuridine) will be assessed pre- and post-procedure for any sibling opting for liver transplantation to assess for treatment response. NCS was offered for the other siblings but they refused the procedure.
The total number of cases of MNGIE reported in the paper is a crude estimate considering cases have been collected from published scientific papers and conference proceedings. This represents a minimum estimate considering many factors - the rarity of presentation and delayed diagnosis in patients, reports of deceased family members with similar clinical presentations who are not counted, undiagnosed cases, and diagnosed but unpublished MNGIE cases worldwide. It has to be stressed that MNGIE-like phenotype affects POLG1, RRM2B, LIG3, RRM1, MTTV1, and MT-RNR1 genes.
Due to the rarity of MNGIE and its complex multisystem clinical picture, patients typically undergo a number of different specialty referrals over many years before obtaining a correct diagnosis. 40 Furthermore, due to the progressive nature of the disease, a late diagnosis is often associated with a poor response to therapy and subsequent poor prognosis. 41 Therefore, it is crucial for physicians to be aware that the radiological characteristics and the clinical manifestations are not restricted to the aforementioned ones and that MNGIE cases can have features similar to meningoencephalitis both clinically and radiologically.
Patient perspective
Affected family members feel that they finally have a reason for the symptomology. They lament that no cure is available for the rare/orphan disease. Sibling 2 has tentatively agreed for an orthotopic liver transplantation. However, he express reticence especially given the risk of opportunistic infections, along with immunosuppressant intake and no guarantee of a cure. Despite that, he awaits a suitable donor. They also express difficulty in gaining weight despite trying various approaches. They wish for a permanent cure for the condition and understand that there are experimental approaches for MNGIE. However, they express that erythrocyte-encapsulated thymidine phosphorylase (EE-TP) and platelets transfusion therapies offer temporary benefits with the need to have additional courses of treatment.6,42,43
Supplemental Material
Supplemental Material - Meningoencephalitis in a novel mutation in MNGIE (mitochondrial neurogastrointestinal encephalomyopathy) ending a familial diagnostic odyssey: A case series report
Supplemental Material for Meningoencephalitis in a novel mutation in MNGIE (mitochondrial neurogastrointestinal encephalomyopathy) ending a familial diagnostic odyssey: A case series report in Odawara by Noor Redha, Zahra Al-Sahlawi, Hasan Hasan, Tejal Shamik Shah, Sara Ghareeb, and Hani Humaidan in Journal of Central Nervous System Disease.
Ethical statement
Ethical approval
The study has been approved by the research committee for Government Hospitals Bahrain (approval serial number 63060623).
Informed consent
Written consent has been obtained from extant patients and from next of kin in deceased patients.
Footnotes
Author contributions
Noor Abdulla Redha: Conceptualization, Investigation, Project administration, Resources, Writing – original draft, Writing – review & editing, Zahra Alsahlawi: Data curation, Formal analysis, Investigation, Writing – original draft, Writing – review & editing, Hasan Hasan: Conceptualization, Data curation, Formal analysis, Visualization, Writing – original draft, Tejal Shamik Shah: Resources, Writing – review & editing, Sara Alghareeb: Resources, Writing – review & editing, Hani Humaidan: Investigation, Writing – review & editing.
Declaration of conflicting interests:
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding:
The author(s) received no financial support for the research, authorship, and/or publication of this article.
Supplemental Material:
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.