
Abstract
The role of uptake transporter (organic anion–transporting polypeptide [Oatp]) in the disposition of a P-glycoprotein (P-gp) substrate (digoxin) at the barriers of central nervous system, namely, the blood-brain barrier (BBB), blood-spinal cord barrier (BSCB), and brain-cerebrospinal fluid barrier (BCSFB), was studied using rat as a preclinical species. In vivo chemical inhibition of P-gp and Oatp was achieved using elacridar and rifampicin, respectively. Our findings show that (1) digoxin had a low brain-to-plasma concentration ratio (B/P) (0.07) in rat; (2) in the presence of elacridar, the B/P of digoxin increased by about 12-fold; (3) rifampicin administration alone did not change the digoxin B/P significantly when compared with digoxin B/P alone; (4) rifampicin administration along with elacridar resulted only in 6-fold increase in the B/P of digoxin; (5) similar fold changes and trends were seen with the spinal cord-to-plasma concentration ratio of digoxin, indicating the similarity between BBB and the BSCB; and (6) unlike BBB and BSCB, the presence of rifampicin further increased the cerebrospinal fluid-to-plasma concentration ratio (CSF/P) for digoxin, suggesting a differential orientation of the uptake transporters at the BCSFB (CSF to blood) compared with the BBB (blood to brain). The observations for digoxin uptake, at least at the BBB and the BSCB, advocate the importance of uptake transporters (Oatps). However, the activity of such uptake transporters became evident only after inhibition of the efflux transporter (P-gp).
Keywords
Introduction
The presence of a distinct barrier between the blood stream and the central nervous system (CNS) compartments is evident from the differential distribution of numerous compounds between the blood-brain and blood-peripheral compartments. The blood-brain barrier (BBB) is a major determinant factor in the successful treatment of brain diseases and is attributed to the distinct morphology of the neurovascular unit that restricts the passage of several drugs to the brain.1,2 The protective mechanism of the CNS barrier acts in preventing the passage of toxic drugs and endobiotics to brain. But the same protective mechanism results in subefficacious drug delivery to the brain and may lead to poor treatment of several brain diseases. The role of efflux transporters, especially P-glycoprotein (P-gp), is highly acknowledged in the constrained delivery of numerous drugs to the brain. It has been observed that in preclinical species, the expression of P-gp is greater than other efflux and uptake transporters in the brain capillaries.3,4 However, the brain vasculature also quarters several other transporters, including uptake transporters that ferry the drug across the barrier to the brain parenchyma.5,6 There are limited studies which have actually reported the role of uptake transporters in transporting drugs across the BBB.7,8 The substrate overlap between P-gp/other efflux transporters and uptake transporters (eg, organic anion–transporting polypeptide [OATP/Oatp]) makes it difficult to segregate between the efflux and uptake of drugs across the barrier unless very specific substrates and inhibitors are used.7–9
Comparison across human OATPs and rodent Oatps has indicated that there exists for the most part similar tissue localization for both. 5 Studies have implicated the localization of an Oatp in both the luminal and the abluminal membranes of the mouse brain capillary endothelial cells. 5 Organic anion–transporting polypeptides have been demonstrated to be important for passage of certain drugs across the BBB. 10 The presence of Oatps in the blood-spinal cord barrier (BSCB) is not widely reported although it has been shown that the efflux transporter (P-gp) localization is similar between the BBB and BSCB. 11 Hence, many a times it has been assumed that the BBB and the BSCB have similar pattern of the transporter expression and localization. The study of BSCB transporters especially becomes important when the site of drug action specifically resides in the spinal cord (eg, certain pain disorders).11,12 There is limited literature describing the presence and localization of transporters at the blood-cerebrospinal fluid barrier (BCSFB).13,14 The P-gp is localized on the BCSFB, but it has been suggested that the orientation of the efflux transporter may enable the transport from the blood to the CSF instead of effluxing drugs out of the CSF.14,15
Although there is substrate overlap between rodents and human OATP/Oatps, transporter function shows significant species differences in capacity. In vitro studies indicate that the hepatic Oatp transporter activity is more prominent in rodents compared with humans. 16 Many Oatp substrates also show overlapping substrate specificity for P-gp and other efflux transporters, such as breast cancer resistance protein (BCRP) or multidrug resistance–associated proteins (MRPs). Digoxin is a widely used P-gp substrate in in vitro as well as in vivo preclinical and clinical studies, and its disposition is influenced by P-gp modulation across various membranes, including the BBB. Digoxin brain penetration increases ~10-fold when P-gp activity is inhibited either by chemical inhibition or by P-gp gene knockout in preclinical species.17,18 Digoxin is also a substrate of human and rodent OATP/Oatps, and hepatic and renal uptake of digoxin is mediated by uptake transporters.5,19 Sugiyama et al 5 have shown using the in situ brain perfusion technique that digoxin uptake in the mouse brain was reduced in the absence of Oatp at the BBB. 5 Ose et al 20 reported using Oatp knockout mouse that the brain uptake of digoxin was significantly reduced in Oatp1a4(−/−) mice only when the brain efflux function of P-gp was inhibited by elacridar. 20 Organic anion–transporting polypeptide transporter activity and its sensitivity to P-gp modulation at the rat BBB have not been well characterized. In this study, we have tried to characterize the role of uptake transporters in the digoxin transport across various barriers of CNS. Rifampicin, a pan-inhibitor of several uptake transporters including the OATPs, was used to study the effect of uptake transporter inhibition at the barriers of the CNS. Single dose of rifampicin is known to have inhibitory effect on transporters such as OATPs, whereas chronic dosing rifampicin is shown to have an induction effect on certain enzymes and transporters. Similarly, codosing of rifampicin and digoxin orally has shown to reduce absorption of digoxin due to rifampicin-mediated P-gp induction. 21
The clearance of drugs by metabolism in the brain tissue or endothelia and distribution by bulk flow are considered to be negligible for most of the drugs. 13 Hence, the interplay between passive diffusion, uptake transporters, and efflux transporters determines the passage of a drug across the BBB. Here, we speculate that the presence of highly competent efflux transport renders the uptake transport and passive diffusion insignificant, and hence, a drug like digoxin exhibits poor permeability. We hypothesize that the effect of active uptake transport is masked when efflux transport is dynamically more active (higher affinity and higher capacity). To test this hypothesis at the rat CNS barriers, we have studied the uptake of digoxin at different barriers of the CNS in the presence and absence of P-gp and/or Oatp activity. Our protocol can be implemented to get a mechanistic understanding of the passage across the BBB for compounds which have low to moderate passive permeability and are efflux as well as uptake transporter substrates. The use of chemical inhibition instead of gene knockout animals provides an additional cost benefit.
Materials and Methods
Chemicals
Rifampicin (>95% purity) and digoxin (>98% purity) were purchased from Sigma-Aldrich (Sigma-Aldrich Chemie, GmbH, Munich, Germany). Elacridar (>98% purity) was synthesized by the Discovery Chemical Synthesis department at Biocon Bristol Myers-Squibb Research and Development Center (BBRC), Bangalore, India. Polyethylene glycol 400 (PEG-400) was purchased from Sigma-Aldrich (Saint Louis, MO, USA). Citric acid monohydrate and citric acid trisodium salt dihydrate for the citrate buffer were obtained from Amresco (Solon, OH, USA). Hydroxypropyl-β cyclodextrin (HPβCD) and dimethylacetamide (DMAc) were from Roquette (Lestrem, France) and Merck (Darmstadt, Germany), respectively. MultiScreen Solvinert filter plates (0.45 µm, low-binding hydrophilic polytetrafluoroethylene) were purchased from Millipore (Carrigtwohill, Ireland). High-performance liquid chromatography–grade methanol was purchased from Merck (Mumbai, India). Formic acid was purchased from Fluka (Fluka Chemie, GmbH, Munich, Germany) and Milli-Q water from Milli-Q system (Millipore SAS, Molsheim, France). Polyethylene-50 tubing was purchased from Smiths Medical (ASD incorporation, Dublin, OH, USA), and 22-gauge needles were purchased from Becton Dickinson India Pvt Ltd (Bangalore, India).
Animals
Male Sprague Dawley rats weighing 300 to 350 g (10-12 weeks of age) were obtained from Syngene in-house breeding facility, Bangalore (India). All animal experiments were conducted in an animal research facility of Syngene International Ltd., Bangalore, India, which was registered by the Committee for the Purpose of Control and Supervision on Experiments on Animals and accredited by Association for Assessment and Accreditation of Laboratory Animal Care International, after getting approval of the Institutional Animal Ethics Committee. The animals were fed with a standard laboratory rodent diet (Tetragon Chemie Pvt. Ltd., Bangalore, India) and housed at room temperature (22 ± 3°C) and relative humidity of 50 ± 20% on a 12-hour light and dark cycle. Water was provided ad libitum throughout the study.
Formulation
An oral solution formulation of rifampicin (6 mg/mL) was developed using 10% (v/v) DMAc, 80% (v/v) PEG-400, and 10% (v/v) 0.1 M citrate buffer pH 3.0, based on the physicochemical properties of rifampicin. Elacridar (2.5 mg/mL) and digoxin (1 mg/mL) were coformulated as a solution. 15 Briefly, accurately weighed quantities of elacridar and digoxin were dissolved in DMAc to a concentration of 25 and 10 mg/mL, respectively. To this solution, a mixture containing 1:1.25 ratio of PEG-400 and 60% (w/v) HPβCD in water was added to make up the volume. The final formulation for digoxin and elacridar contained 10% (v/v) DMAc, 40% (v/v) PEG-400, 30% (w/v) HPβCD, and 20% (v/v) water. The solution formulation for combining all the 3 drugs was challenging, so a separate formulation for oral dosing of rifampicin was used.
In vivo study
Rats were kept in a restrainer and the compounds (digoxin alone or digoxin + elacridar) were dosed via tail vein as a bolus at 2 mL/kg dose volume. The doses used were 2 mg/kg for digoxin and 5 mg/kg for elacridar. For the combination study with OATP inhibitor, rifampicin was dosed orally (30 mg/kg, 5 mL/kg) 1 hour prior to the combination dosing of digoxin and elacridar.
The CSF, plasma, spinal cord, and brain samples were collected at 1, 3, 5, 7, and 24 hours using separate set of 3 rats for each time point. For each group, 15 rats were used, of which 3 rats were used for each sampling time point. After isoflurane anesthesia, blood was collected through retro-orbital plexus in tubes containing 2% w/v potassium EDTA solution, and CSF was collected by puncturing cisterna magna. 15 Immediately after blood and CSF collection, rats were euthanized using carbon dioxide; brain and spinal cord were harvested and homogenized using 4 volumes of water. The CSF, plasma, and cord and brain homogenate samples were stored at −80°C until further analysis.
Standard solutions
Primary stock solutions of digoxin, elacridar, rifampicin, and alprazolam (internal standard [IS]) were prepared at 2 mM and stored at 4°C. Calibration standards were prepared in serial dilution mode from 10 000 to 1.22 nM in rat blank plasma on the day of analysis. The lower limit of quantitation for digoxin, elacridar, and rifampicin were 2.44, 1.22, and 1.22 nM, respectively. The assay accuracy was found to be between 85% and 115% to that of nominal concentration.
Sample preparation method (plasma, CSF, and spinal cord and brain homogenates)
Aliquots of standards and study samples (25 µL) were quenched with 125 µL of acetonitrile (containing 150 nM alprazolam as an IS) in 96-well hydrophilic solvinert plates (Millipore Corporation, Darmstadt, Germany, Cat. No. : MSRP N04 50). The plates were vortexed on a plate shaker for 5 minutes at 300 rpm and then centrifuged at 4000g for 5 minutes. The concentrations of test compounds (digoxin, elacridar, and rifampicin) in the filtrate were determined by ultra-performance liquid chromatography-tandem mass spectrometry (UPLC-MS/MS). Calibration curves were prepared using blank rat plasma and artificial CSF. Brain and spinal cord samples were analyzed for digoxin, elacridar, and rifampicin using the rat plasma calibration curve after a 5-fold dilution of the brain/spinal cord homogenates.
Instrumentation and chromatographic conditions
Waters Acquity UPLC Integrated System (Miliford, USA) was used to inject 3-µL aliquots of the processed samples on a reverse phase column (Waters: BEH C18, 1.7 µm, 2.1 mm × 50 mm) which was maintained at 40 ± 2°C in a column oven. The system was run in gradient mode at a flow rate of 0.6 mL/min with mobile phase A containing 0.1% formic acid in water and mobile phase B containing 0.1% formic acid in acetonitrile.
Quantitation was achieved by MS/MS detection in positive ion, multiple reaction monitoring mode for analytes (digoxin, elacridar, and rifampicin) and IS (alprazolam) using API 4000 Q-Trap mass spectrometer (Applied Biosystems, MDS Sciex, Toronto, ON, Canada) equipped with an API electrospray ionization source. Alprazolam was selected as IS based on the performance comparing the mass response, stability, and retention time with the analytes. The source parameters were optimized to obtain stable response for analytes, namely, temperature of 550°C and 5500 V ion spray voltage; curtain gas, nebulizer (GS1), and auxiliary gases (GS2) were set at 30, 45, and 50 psi, respectively. Quadrupoles Q1 and Q3 were set on unit resolution, and the dwell time set for each Selected Reaction Monitoring (SRM) was 30 ms. The transition pairs of Q1/Q3 used for digoxin, elacridar, rifampicin, and alprazolam were 798.4/651.2, 564.2/252.2, 823.5/791.3, and 309/281, respectively. The analytical data were processed by Analyst software (version 1.5.0).
Pharmacokinetic analysis
Area under the concentration-time profile (AUC) was calculated by noncompartmental analysis and mixed log linear method using Kinetica software (version 4.4.1; Thermo Electron Corporation, Waltham, MA, USA). Brain-to-plasma concentration ratio (Kp, Brain ) was calculated using the ratio of digoxin concentration in the brain and plasma at each time point. Partition coefficients (Kp, AUC ) for brain, spinal cord, and CSF were calculated using the ratio of the AUCs obtained from the concentration-time profile in the respective matrix relative to plasma:

Statistical analysis
Statistical analysis was performed using the GraphPad Prism program version 5.02 (GraphPad software, San Diego, CA, USA). Student t test was conducted to compare the brain-to-plasma concentration ratio (Kp,Brain) of digoxin obtained at each time point (1, 3, 5, and 7 hours) when administered alone and in combination with elacridar and/or rifampicin; P < .05 was regarded as significant.
Results
Role of uptake transporters at the BBB
Digoxin showed low brain penetration as evident from the KP,AUC, Brain of ~0.07 when administered at 2 mg/kg, intravenously (Figure 1A). It is a well-known substrate of P-gp, and in the presence of a P-gp inhibitor, elacridar (5 mg/kg, intravenously), the brain-to-plasma ratio increased ~13-fold (KP,AUC, Brain = 0.89). Furthermore, in the presence of P-gp inhibition, we sought to mine the role of uptake transporters by administering rifampicin (30 mg/kg, oral). The KP,AUC, Brain for digoxin increased from 0.07 to 0.42 (~6-fold) in the presence of both elacridar and rifampicin. Thus, the KP,AUC, Brain reduced by 2-fold from 0.89 to 0.42 in the presence of an OATP inhibitor, rifampicin, when added on top of elacridar.
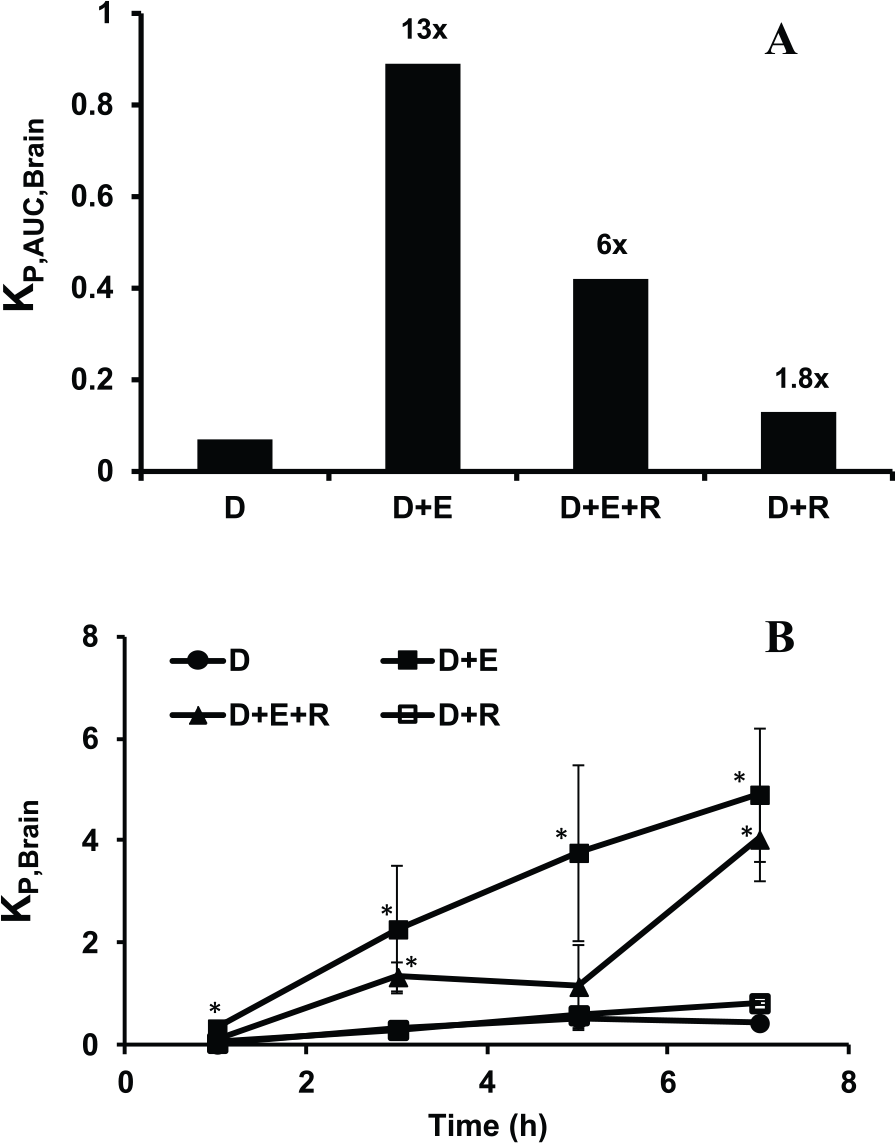
(A) Comparison of KP,AUC, Brain of digoxin when administered alone and in combination with elacridar and/or rifampicin. The numbers above each bar indicate the fold increase in KP,AUC, Brain compared with digoxin alone. For each group, 15 rats were used, of which 3 rats were used for each sampling time point. As the KP,AUC, Brain was calculated using the KP, Brain at each time point collected from different sets of rats, error bars were not included. (B) Comparison of KP, Brain of digoxin at each time when administered alone and in combination with elacridar and/or rifampicin. Concentrations represent mean ± SD for each time point (n = 3). *P < .05 by t test by comparing the concentrations of each time point with the concentrations of the respective time point when digoxin was administered alone. D indicates digoxin; E, elacridar; R, rifampicin.
Rifampicin alone was not effective in altering digoxin uptake across the BBB (KP,AUC, Brain = 0.13). The difference in the uptake of digoxin in the presence of rifampicin was less than 2-fold as with digoxin uptake without rifampicin, and the poor brain uptake was still evident due to the presence of P-gp in rats. This suggests that the role of uptake transporters could only be evident when P-gp was inhibited. Figure 1B shows the KP, Brain of digoxin at different time points when administered alone and in combination. The presence of rifampicin when P-gp was inhibited by elacridar clearly showed reduction in KP.
Figure 2 shows the unbound concentration-time profiles of elacridar and rifampicin. The unbound concentration of elacridar in plasma was higher (>10-fold) than its inhibitory concentration (Ki) for P-gp (1.6 nM) 17 at its peak concentration, and the levels were maintained above the Ki for ~15 hours. Thus, the dose of elacridar (5 mg/kg) used in this study showed sufficient plasma exposure to inhibit P-gp at the BBB. The Ki of rifampicin for the uptake transporters (OATP1B1) is reported to be 1.1 µM. 22 In this study, the maximum unbound concentration of rifampicin (1.8 µM) was higher than its Ki, and the level was maintained above Ki for up to ~5 hours, which may be sufficient for the inhibition of Oatp. Rifampicin is reported to be a pan-inhibitor of the uptake transporters, and the concentrations achieved in this study may be sufficient enough to demonstrate uptake transporter inhibition at the BBB. The role of rifampicin in inhibiting the specific uptake transporter at the BBB needs to be evaluated. However, with the pan-Oatp inhibitory properties of rifampicin, it is evident that it inhibits uptake transporters, especially Oatps, at different locations in the body including the BBB.

Unbound concentration-time profile of (A) elacridar and (B) rifampicin in plasma. D indicates digoxin; E, elacridar; P-gp, P-glycoprotein; R, rifampicin.
Role of uptake transporters at the BSCB
To observe the role of transporters at the BSCB, the spinal cord was harvested and the KP,AUC, Spinal cord was determined. The trend of digoxin transport across the BSCB was similar to that observed at the BBB (Figure 3). Digoxin uptake was low in the absence of any drug transporter inhibitors (KP,AUC, Spinal cord = 0.06). In the presence of elacridar, the spinal cord uptake of digoxin increased by ~10-fold (KP,AUC, Spinal cord = 0.62) and is thus evident of the role of efflux transport of P-gp at the BSCB similar to the BBB. Furthermore, in the presence of both elacridar and rifampicin, although the spinal cord-to-plasma digoxin ratio increased by ~6-fold (KP,AUC, Spinal cord = 0.37), it was lower compared with the digoxin BSCB uptake in the presence of elacridar alone. Similar to the BBB, the increase in the digoxin uptake at the BSCB in the presence of rifampicin alone was less than 2-fold compared with digoxin alone.

Comparison of KP,AUC, Spinal cord of digoxin when administered alone and in combination with elacridar and/or rifampicin. The numbers above each bar indicate the fold increase in KP,AUC, Spinal cord compared with digoxin alone. For each group, 15 rats were used, of which 3 rats were used for each sampling time point. As the KP,AUC, Spinal cord was calculated using the KP, Spinal cord at each time point collected from different sets of rats, error bars were not included. D indicates digoxin; E, elacridar; R, rifampicin.
Role of uptake transporters at the BCSFB
The uptake of digoxin at the BCSFB followed a different pattern in comparison with the BBB and the BSCB (Figure 4). The digoxin KP,AUC, CSF without any inhibitors was 0.02, and in the presence of elacridar, it increased by ~3-fold to 0.06. The extent of increase in digoxin uptake at BCSFB was found to be lower than the uptake observed at BBB (13-fold) and BSCB (10-fold) in the presence of elacridar, which indicates differential orientation of P-gp at the BCSFB.

Comparison of KP,AUC, CSF of digoxin when administered alone and in combination with elacridar and/or rifampicin. The numbers above each bar indicate the fold increase in KP,AUC, CSF compared with digoxin alone. For each group, 15 rats were used, of which 3 rats were used for each sampling time point. As the KP,AUC, CSF was calculated using the KP,CSF at each time point collected from different sets of rats, error bars were not included. D indicates digoxin; E, elacridar; R, rifampicin.
In the presence of both elacridar and rifampicin, digoxin uptake (KP,AUC, CSF ) at the BCSFB increased to 0.13 (~7-fold) compared with digoxin alone. This increase in digoxin uptake by inhibiting both P-gp and Oatp was less than 2-fold higher than by inhibiting P-gp alone. Furthermore, by inhibiting only Oatp by administering rifampicin, the digoxin uptake (KP,AUC, CSF = 0.08) was ~4-fold greater compared with digoxin uptake without any transporter inhibition at the BCSFB. The different patterns of digoxin uptake in the presence and absence of individual or dual transporter inhibition suggest differential orientation of efflux and uptake transporters at the BCSFB compared with the BBB and BSCB. 15 As opposed to the BBB and BSCB, the role of uptake transporters was evident even in the presence of efflux (P-gp), suggesting that at the BCSFB the direction of transport of the Oatps is mainly from the CSF to the blood side.
Discussion
The role of P-gp in the passage of digoxin across biological membranes has been well elucidated. 23 The presence of P-gp results in low brain permeability of digoxin, and on P-gp inhibition, the brain uptake of digoxin was increased by approximately 10-fold. Digoxin is also a known substrate for uptake transporters, and the importance of these transporters for digoxin uptake in the liver and kidney has been demonstrated in rats.5,19,24 There are, however, contradictory reports with respect to the interaction and substrate specificity of digoxin with Oatps.25,26 Using an in vivo protocol, we have tried to look into the possible role of the uptake transporters such as the Oatps in influencing the digoxin uptake at the rat BBB. Due to the overwhelming activity of P-gp, the effect of Oatps at the BBB is difficult to assess as both the transporters are accessible to the drug at the luminal side of the endothelial barrier. This orientation is in contrast to the orientation of uptake and efflux transporters in the liver. In the liver, the uptake transporters such as Oatps, Oct (organic cation transporter), and Ntcp (sodium/taurocholate cotransporting polypeptide) are present at the sinusoidal flank, whereas the efflux transporters such as P-gp, Bcrp, Mrp2, and Bsep (bile sale export pump) are present at the canalicular brim. Similarly, in renal tubular cells, the uptake transporters, predominantly the Oatps and Octs, are present facing the blood side and the efflux transporters, such as P-gp and Mrps, are present at the luminal margin. The BBB is similar to the intestinal epithelia in that drugs are exposed to the uptake as well as efflux transporters at the same time.
In this study, we have used elacridar as a tool to inhibit P-gp, and in its presence, digoxin uptake across the BBB was increased by about ~13-fold. The unbound concentration of elacridar in plasma (Iu/Ki > 1 for ~15 hours) was sufficient enough to inhibit P-gp at CNS barriers. Rifampicin was used as an Oatp inhibitor at a tolerable dose of 30 mg/kg orally. The concentration of rifampicin in the systemic circulation when given along with elacridar was sufficient (Iu/IC50 > 1 for ~5 hours) to inhibit the uptake transporters and also suggested by the decrease in digoxin uptake when compared with the digoxin uptake in the presence of P-gp inhibition alone. In the presence of rifampicin alone, digoxin uptake across the BBB remained low. For drugs with low passive permeability, carrier-mediated uptake transport could be demonstrated only after inhibiting the efflux, which is due to the high abundance and activity of the efflux transporters, especially P-gp. For many drugs, such high brain uptake is observed after P-gp and/or Bcrp inhibition. There is always a coexistence of passive permeability and carrier-mediated transport, with one of the processes playing a more prominent role than the other. For high passively permeable compounds (quinidine, verapamil), in the absence of efflux, the activity of uptake transporters is overwhelmed by passive diffusion resulting in no effect of inhibitor on uptake transporters. This study offers an effective tool to realize the more prominent process or coexistence of 2 processes in the absence of efflux transport. Recent studies have demonstrated a substantial increase in brain uptake of drugs when P-gp-Bcrp are knocked out together as opposed to single P-gp or Bcrp knockout.27–30 In such cases, it would be interesting to check the importance of uptake transporters after all the efflux liabilities have been knocked out.
The results also suggest that further knowledge of uptake transporters can be capitalized to enhance drug delivery across the CNS barriers. Medicinal chemists could work around the Structure Activity Relationship (SAR) to reduce efflux, incorporate uptake transporter substrate features, and increase permeability across the CNS barriers.31,32 Chemical inhibition of efflux transporters at the BBB cannot be achieved clinically and hence not a viable strategy for CNS drug delivery. 33 It has been shown in diseases such as neuropathic pain that uptake transporters are overexpressed at the BBB, and such knowledge could be useful in targeting a drug across the CNS barriers by incorporating uptake transporter features into the drug design.12,34,35 Organic anion–transporting polypeptide are also implicated to be overexpressed in certain cancers and are being targeted for transporter-mediated drug delivery. 36 However, the impact of overexpression of uptake transporters on the extent of reduction in masking the uptake by P-gp needs to be investigated. OATP1A2 is expressed at the luminal membrane of the human BBB, and oatp1a4 is expressed at the rat BBB. Rat oatp1a4 is thought to be one of the homologs of human OATP1A2. 37 Such human uptake transporter homologs at the rat brain may render the rat as a relevant model to investigate the role of uptake transporters in the human CNS system. Rifampicin inhibits the uptake transporters, especially various OATPs/Oatps, and should be further evaluated to recognize its potency against the various OATPS/Oatps especially at the barriers of the CNS.
Our study has also indicated that the transporter expressions at the BBB and the BSCB are identical, whereas there is difference in the transporter expression and/or transport direction for the BCSFB compared with the other 2 barriers (Figure 4). Further characterization of uptake transporters is warranted to understand their role in the CNS uptake of drugs that might be masked by the overwhelming activity of an efflux transporter. Examples of such drugs include taxanes, statins, and tyrosine kinase inhibitors that are substrates of both efflux and uptake transporters.27–30,38–41
Footnotes
Acknowledgements
The authors thank Jadiswami Channayya for helping in the animal studies.
Peer Review:
Five peer reviewers contributed to the peer review report. Reviewers’ reports totaled 1396 words, excluding any confidential comments to the academic editor.
Funding:
The author(s) received no financial support for the research, authorship, and/or publication of this article.
Declaration of Conflicting Interests:
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Author Contributions
KST and TTM conceived and designed the experiments, wrote the first draft of the manuscript, and jointly developed the structure and arguments for the paper. KST, VK, RM, KR, and TTM analyzed the data. KST, VK, SG, SKS, PM, SM, and TTM contributed to the writing of the manuscript. KST, PM, SM, and TTM agree with manuscript results and conclusions, made critical revisions, and approved the final version. All authors reviewed and approved the final manuscript.